
H-ZSM-5催化甲苯與碳酸二甲酯和甲醇甲基化的反應(yīng)機理
2013-09-17 06:58:34李玲玲JANIKMichael聶小娃宋春山郭新聞
物理化學學報 2013年7期
李玲玲 JANIK J.Michael 聶小娃,4 宋春山,2,3 郭新聞,*
(1大連理工大學化工學院精細化工國家重點實驗室,PSU-DUT聯(lián)合能源研究中心,遼寧大連116024;2賓夕法尼亞州立大學能源與礦物工程系能源研究所,PSU-DUT聯(lián)合能源研究中心,賓夕法尼亞16802,美國;3賓夕法尼亞州立大學化學工程系,賓夕法尼亞16802,美國;4俄亥俄州立大學化工與生物分子工程系,俄亥俄43210,美國)
1 Introduction
Para-xylene,an important petrochemical,can be produced from multiple processes including isomerization of xylenes and methylation of toluene.ZSM-5,a solid zeolite catalyst,shows a greater selectivity in synthesis of para-xylene by toluene methylation than other zeolites.1-3High selectivity of paraxylene has been obtained in toluene methylation with methanol as a traditional methylation reagent.3Recently,the use of dimethyl carbonate(DMC),which is an important green chemical intermediate,has gained attention in xylene production.4,5The methylation of toluene with DMC in a fixed-bed reactor demonstrated positive selectivity to para-xylene over MgO-modified MCM-22.4Toluene methylation with methanol,however,could give higher selectivity to para-xylene than methylation with DMC which has a faster coke deposition rate over Si-P-Mg modified ZSM-5.5
Increasing both the selectivity to para-xylene and the stability of the catalyst remains important issues in catalyst development for toluene methylation.Mechanism determination will facilitate rational design of selective zeolite catalysts for toluene methylation.Experimental kinetic studies show promising results of toluene alkylation with DMC and methanol using a ZSM-5 catalyst,and determination of differences in key steps within the two alkylation processes can aid in interpreting differences in catalytic activity and optimal catalyst design between the two reagents.Herein,we apply density functional theory(DFT)6calculations,using the embedded quantum mechanics/molecular mechanics(QM/MM)approach,to elucidate mechanistic differences between methanol and DMC reagents in the methylation of toluene.
The mechanism of aromatic methylation with DMC over zeolites is not well established.The DMC molecule has three isomer configurations including trans-trans,cis-cis,and trans-cis structures,with the trans-trans being the most stable configuration.7DMC can be decomposed to dimethyl ether,methanol,and carbon dioxide over H-ZSM-5 zeolite,and the dimethyl ether is speculated to be produced by reaction of methoxide and methanol.8-10The decomposition products can further react with toluene.11-15We can speculate the mechanism of toluene methylation with DMC based on these clues.
Aromatic alkylation with methanol as a reagent has received more prior attention.Previous experimental and theoretical research suggests that toluene alkylation with methanol to xylene isomers can occur through either stepwise or concerted paths.11-15The stepwise path passes through a surface methoxide species whereas the concerted path includes a direct alkylation by methanol.The exploration of the dominant reaction path might help guide rational catalyst design to increase the selectivity to para-xylene.Vos et al.15examined the concerted mechanism of toluene methylation with methanol using a periodic model of MOR zeolite.The difference in pore structure and therefore active site structure in highly active ZSM-5 motivates our further mechanistic study of this reaction.
Theoretical calculation based on quantum chemistry is a helpful tool to investigate molecular level catalytic phenomena.The“our own-N-layered integrated molecular orbital+molecular mechanics”(ONIOM)16,17approach with two layers was used herein to examine toluene methylation with DMC and methanol.Equilibrium and transition state configurations were located within the H-ZSM-5 pore,and relative energies of these states provide elementary step activation barriers and reaction energies.The QM region,including the acid site and reacting species,provided an accurate description of reaction energetics and was combined with a MM representation of the confining extended framework.Following B3LYP/UFF optimization,single point energies with the ωB97X-D functional were used to provide accurate zeolite-hydrocarbon interaction energies including dispersion energies.
2 Computational model and methods
A 128T(128 tetrahedra)cluster of H-ZSM-5,including the zigzag and straight channels,was taken from the lattice structure of MFI zeolite and is shown in Fig.1(a,b).18-21A single Al atom is most preferred to substitute for Si at the T12 position to introduce a Br?nsted acid site of ZSM-5.22-25The cluster was terminated by H atoms bound to Si atoms,with the terminal Si-H bond length fixed in the crystalline lattice direction at 0.147 nm.The methods and model used herein are equivalent to that used in our previous study of methylation of 2-methylnaphthalene.18
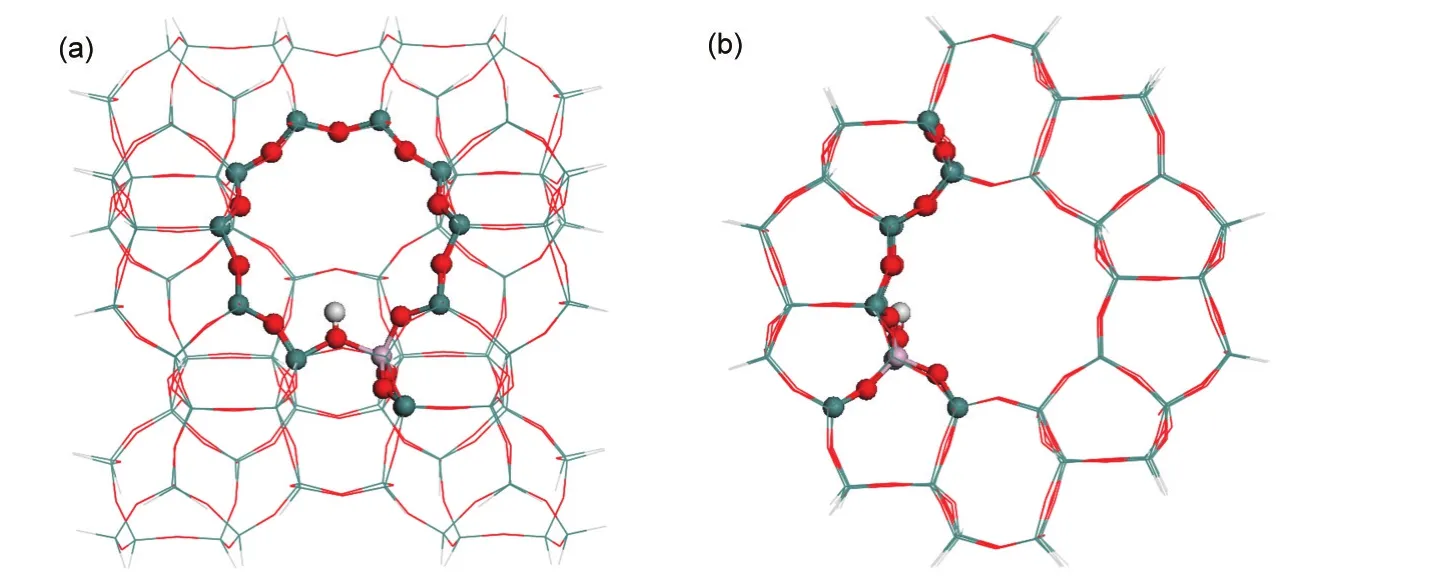
Fig.1 ONIOM2 model of the 128T H-ZSM-5
A two-layer ONIOM(ONIOM2)approach was employed for the 128T cluster.The inner 12T portion,covering a 10-membered-ring(10MR)and two additional basal T units nearby the Al atom,was considered to represent the active region of the zeolite and treated at the B3LYP/6-31G(d,p)level.The remaining extended framework was considered with the universal force field(UFF).26During optimization,only the basic 5T of the active region,[(≡(SiO)3Al(OH)Si≡],and the adsorbates were allowed to relax,whereas the rest of the zeolite model was fixed at the crystallographic coordinates.Structural optimization of the most bulky adsorbed states within the 128T ONIOM cluster was performed considering different QM-portion sizes to test the validity of the 12T QM model.Similar structural parameters and relevant energies determined from the ONIOM clusters with different QM sizes(12T and 27T)demonstrate a reasonable description of stable states using a 12T QM region.The model in Fig.1 is a reasonable balance of accuracy and computational cost considering the bulky reacting species.The quadratic synchronous transit(QST)method within the framework of the ONIOM2(DFT:UFF)model was applied to locate transition states,and each transition state was confirmed to have a single imaginary vibrational frequency.The quasi-intrinsic reaction coordinate(IRC)approach followed geometry optimizations were used to obtain initial reactant and product species toward reaction tendency.27,28
In the ONIOM system,error may be introduced by the coarse combination of the low level and the more accurate DFT method.Based on the definition of Morokuma?s group,29a ΔS-value was used to confirm the reliability of energetics obtained by the level/model combination.A large ΔS-value indicates inaccuracy of the ONIOM2(DFT:UFF)extrapolation.Therefore,after optimization or transition state identification with the ONIOM2 model,a zero point vibrationally corrected single-point energy was calculated using the expensive full ωB97X-D method for the whole 128T cluster.The ωB97X-D functional was developed to consider the binding energies of adsorbates covalently and non-covalently interacting with a zeolite framework.28,30All energetics reported herein refer to the values calculated for the single-point energy using the ωB97XD on the B3LYP(12 T)/UFF(116 T)optimized structure.All calculations were performed with the Gaussian 0331and Gaussian 0932packages.
3 Results and discussion
3.1 Overview of possible reaction mechanisms
Chemical differences between DMC and methanol lead to different reaction mechanisms of toluene methylation.Methanol,as an alcohol,can dehydrate to form a water molecule and a methoxide species which further alkylates toluene,which is generally called the stepwise path.Alternatively,methanol can co-adsorb with toluene and alkylate through a concerted dehydration-methylation reaction.
Based on the intermediates observed in the DMC decomposition reaction8-10and the feasible methylation of toluene with methoxide,11-13a stepwise toluene methylation with DMC may proceed through DMC decomposition and toluene methylation with the surface methoxide species(path A in Scheme 1(a)).A concerted mechanism involving DMC,as speculated by experimental reports,4,33,34partially decomposes DMC to form a methyl group and releases a methyl carbonate(MC)molecule.The methyl group could alkylate toluene in a concerted step,however we found that the decomposed methyl carbenium ion interacts with a zeolite basic oxygen atom instead of directly attacking toluene.The zeolite basic oxygen atom serves as a temporary acceptor.We propose that a“concerted”mechanism of toluene methylation with DMC starts with DMC partial decomposition to form a methyl carbenium ion which interacts with the zeolite medium releasing a MC molecule,then the medium donates the carbenium ion to alkylate toluene as shown by path B and path C in Scheme 1(a).Though we term this as a“concerted”mechanism,the effect of toluene in the initial decomposition step can be ignored to decrease the space restraint and the computational cost.The three paths of toluene methylation with DMC differ in the first decomposition step,forming different DMC-derived intermediate products.
The methylation of toluene with DMC and methanol via the methoxide intermediate is examined in Section 3.2,and the concerted methylation of toluene with methanol is examined in Section 3.3.
3.2 Toluene methylation with DMC and methanol via methoxide intermediate
3.2.1 DMC decomposition
The methylation reaction of toluene with DMC starts with DMC decomposition to form a methoxide intermediate via path A,path B or path C.DMC adsorbs strongly over the Br?nsted acid site of H-ZSM-5 through either bidentate adsorption or unidentate adsorption.35The adsorption structure is preferred for full decomposition or partial decomposition and further methylation with toluene.The optimized structures of key intermediates and transition states during DMC decomposition are shown in Fig.2.In path A of toluene methylation with DMC,DMC fully decomposes to methoxide,methanol,and carbon dioxide,and further the methoxide species then alkylates toluene.In path B or C,DMC partially decomposes to form a methyl carbocation and MC,and the methyl carbocation alkylates toluene.The energetics of key states are given relative to the sum of gas phase DMC and the isolated zeolite in Fig.3.Relevant bond parameters of the key structures are given in Table1.
Fig.3(a)illustrates the reaction energy profile for path A of toluene methylation with DMC,including the full decomposition reaction of DMC to the methoxide intermediate.In the decomposition step,formation of surface methoxide,methanol,and carbon dioxide occurs in a concerted action.Fig.2(a)shows the initial adsorption structure of bidentate DMC which initiates path A of toluene methylation with DMC.DMC adsorbs at the Br?nsted acid site(H1)by formation of a hydrogen bond between H1 and O4,labeled as Ads1_DMC.The O1-H1 bond is elongated from 0.097 nm in the isolated zeolite to 0.099 nm with DMC adsorbed.The DMC molecule is activated after adsorbing,reflected by the lengthening of C3-O4 and C1-O3 bond distances by 0.002 and 0.001 nm,respectively.Catalyzed by H-ZSM-5,DMC completely dissociates through the transition state,TS1_DMC,shown in Fig.2(b).The cleavage of the C3-O4 bond,motivated by H1 attack,promotes the breaking of the C1-O3 bond due to instability of the negative CH3OCO-group.At the transition state,a close-to-planar methyl group suggests a carbenium-like transition state,with the positive charge stabilized by interaction with both the O2 zeolite atom and the O3 atom of what will be the CO2decomposition product.The bond angle of OCO is 172.3°,which shows a tendency towards the generation of a CO2molecule.In the product structure,Int1(Fig.2(c)),the surface methoxide forms a covalent interaction between C1 and O2 at the interatomic distance of 0.150 nm.The formed methanol molecule adsorbs at the framework basic O1 atom by forming a hydrogen bond between H1 and O1.The carbon dioxide molecule is co-stabilized by methoxide(C1…O3)and methanol(C3…O4).
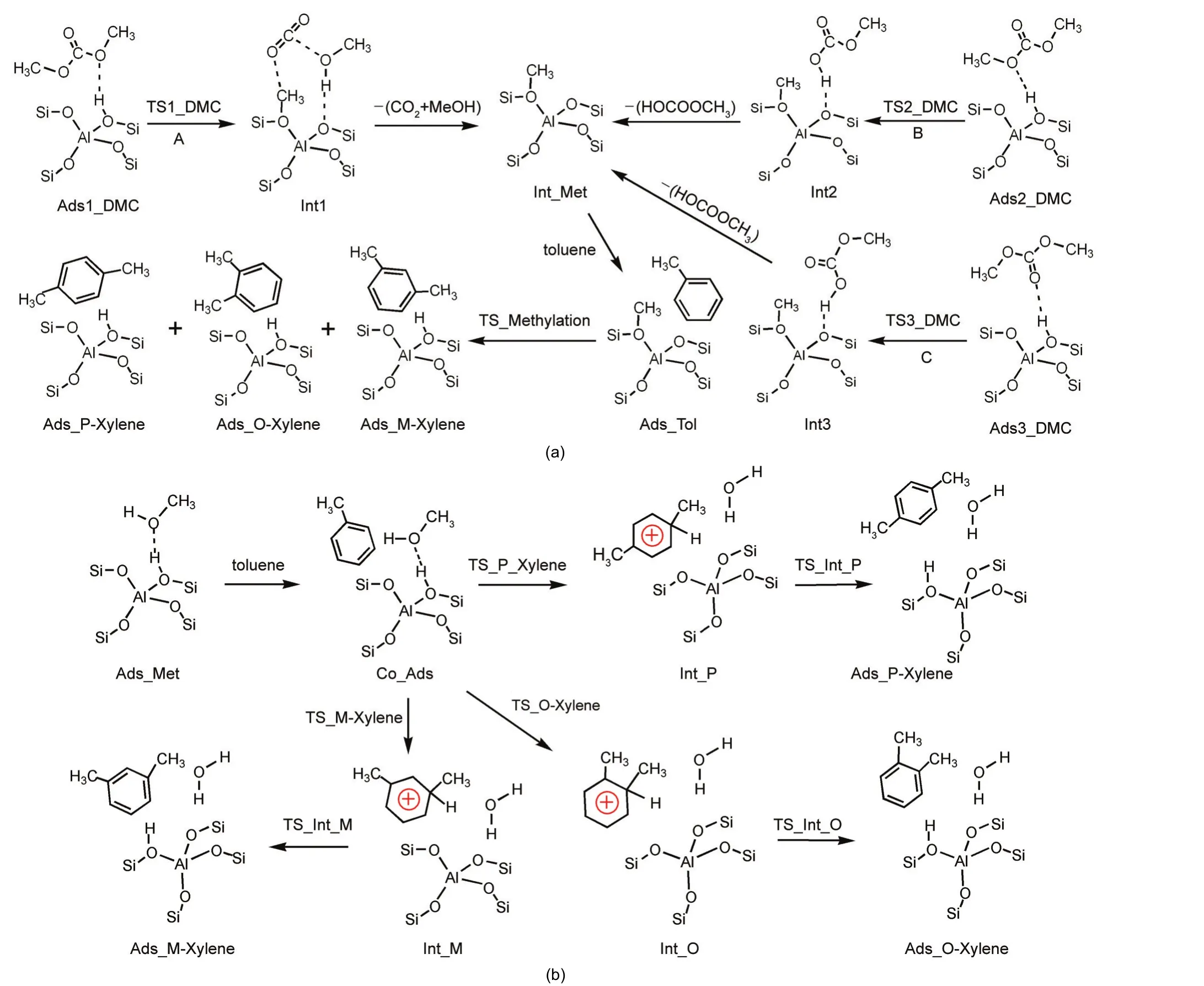
Scheme 1 Proposed reaction paths for methylation of toluene with DMC and methanol over H-ZSM-5
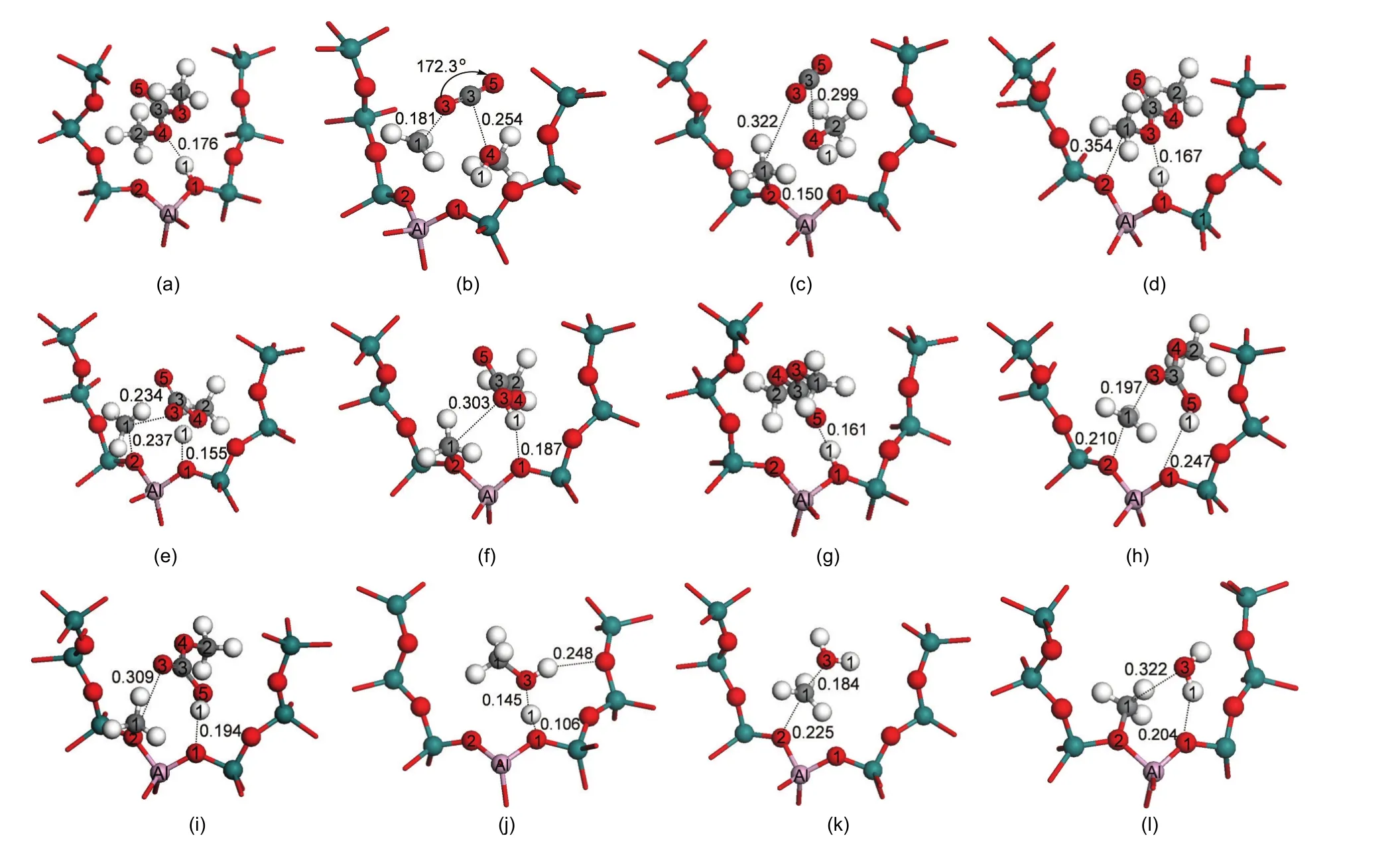
Fig.2 Optimized structures of equilibrium and transition states along the paths of DMC decomposition and methanol dehydration
The adsorption energy of Ads1_DMC is-157.7 kJ·mol-1.This exothermic value is relatively similar to the dimethyl ether adsorption energy(-123.0 kJ·mol-1)19computed with ONIOM2(M06-2X/6-311+G(2df,2p):UFF)+SCREEP calculation.The activation energy(Ea)of the DMC full decomposition step to methoxide,methanol,and carbon dioxide is 186.6 kJ·mol-1catalyzed by H-ZSM-5,which is 28.4 kJ·mol-1higher than that calculated for the same reaction catalyzed by dicylohexylurea in the gas phase.36The produced methanol and car-bon dioxide diffuse from the active site by overcoming a collective desorption energy of 125.1 kJ·mol-1.The produced methanol can diffuse to another Br?nsted active site to participate in a further methylation reaction with a toluene molecule.
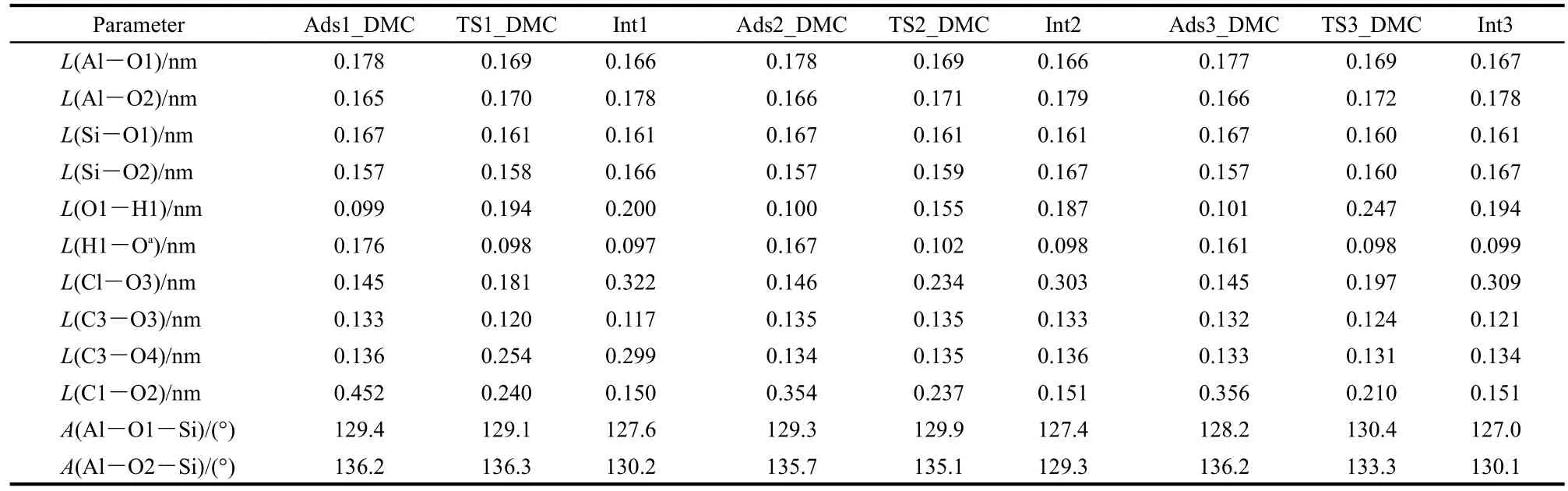
Table 1 Optimized geometric parameters of species involved in decomposition of DMC over H-ZSM-5
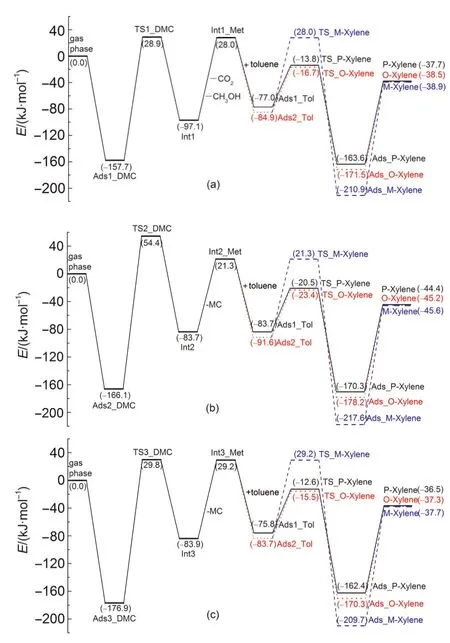
Fig.3 Reaction energy profile for methylation of toluene with DMC to form para-,ortho-,and meta-xylene
Fig.3(b)and 3(c)illustrate the reaction energy profiles for DMC partially decomposing to a methoxide species and a MC molecule in path B and path C of toluene methylation with DMC,respectively.Fig.2(d)displays the initial adsorption structure of bidentate DMC which initiates path B of toluene methylation with DMC.DMC adsorbs at the Br?nsted acid site of H-ZSM-5 through the formation of a hydrogen bond between H1 and O3.In the adsorption structure,Ads2_DMC,the C1-O3 bond of DMC is weakened,as reflected by the expansion of the C1-O3 bond length from 0.144 to 0.146 nm.In the transition state shown in Fig.2(e),TS2_DMC,H1 transfers from the initial basic O1 to O3 atom,and the C1-O3 bond is completely broken to form a methyl carbocation.A methoxide and a MC molecule(Fig.2(f),Int2)are produced via the transition state.To initiate path C,DMC adsorbs at the Br?nsted acid site of the H-ZSM-5 catalyst by the O5 atom,forming a unidentate adsorption structure(Fig.2(g)).This structure is labeled as Ads3_DMC.The bond distance of C1-O3 is lengthened by 0.001 nm,illustrating a tendency to dissociate.In the transition state(Fig.2(h),TS3_DMC),the H-ZSM-5 catalyst donates the H1 proton to bond with the O5 atom,and the C1-O3 bond is motivated to simultaneously rupture.This decomposition step ends by releasing a MC molecule adsorbing in the vicinity of methoxide(Fig.2(i),Int3).
The adsorption energies of Ads2_DMC and Ads3_DMC are-166.1 and-176.9 kJ·mol-1,respectively.The Eafor DMC partial decomposition to methoxide and MC in path B and path C are 220.5 and 206.7 kJ·mol-1,respectively.The formed MC intermediates desorb from the active site overcoming 105.0 and 113.1 kJ·mol-1desorption energies in the corresponding paths.The MC intermediates in path B and path C are isomers.After desorbing from the active site,the MC in path C could transform to the more stable MC produced in path B.The MC is unstable and may quickly dissociate to methanol and carbon dioxide over another Br?nsted acid site of H-ZSM5.Catalyzed by H-ZSM-5,Eafor MC dissociation is 41.8 kJ·mol-1,which is 21.4 kJ·mol-1lower than the calculated barrier reported for gasphase decomposition and 6.7 kJ·mol-1lower than the barrier reported using a solvent model.33Path C is more preferred for DMC to partially decompose to methoxide and MC than path B due to a lower Ea(13.8 kJ·mol-1).However,DMC partial decomposition to form methoxide and MC via path C(Ea=206.7 kJ·mol-1)needs to overcome a higher Eathan full decomposition to form methoxide,methanol,and carbon dioxide via path A(Ea=186.6 kJ·mol-1).
3.2.2 Methanol dehydration
The stepwise path of toluene methylation with methanol resembles toluene methylation with DMC,with the difference being that the methoxide intermediate forms from methanol dehydration.The energy profile for methanol dehydration is given in Fig.4.A methanol molecule adsorbs on the Br?nsted acid site,dehydrates to methoxide,and then the methoxide attacks toluene to form xylene isomers.The methanol is adsorbed on the acid site with two hydrogen bonds between O3 and H1 and between Hmethanoland Oframeworkforming a side-on adsorbed structure reported by Ivanova and Corma,11as shown in Fig.2(j).The adsorption energy is-128.4 kJ·mol-1which is within range of the data calculated by Mazar et al.37by PBE+D method(-118 kJ·mol-1in sinusoidal channel and-132 kJ·mol-1in straight channel),and agrees well with the experimental value((-115±5)kJ·mol-1)38.To form a methoxide,the C1-O3 bond dissociates as H1 is transferred to O3,releasing a water molecule and forming a methyl-carbenium transition state(Fig.2(k)).The methyl-carbenium ion is stabilized by O3 and O2 framework oxygen atoms.The Eaof methanol dehydration to methoxide is 120.9 kJ·mol-1.The methyl group binds to a framework oxygen atom(O2)to form the surface methoxide intermediate(Fig.2(l)).The desorption energy of the produced water molecule is 47.2 kJ·mol-1.We explicitly define the stepwise path to require the desorption of water,as the lack of sta-bilization of the alkylation transition state by a water molecule differentiates the stepwise and concerted transition states.Key bond distances within the methanol dehydration sequence are shown in Fig.2.

Fig.4 Reaction energy profile for stepwise mechanism of methylation of toluene with methanol to form the para-,ortho-,and meta-xylene including methanol dehydration step and methylation step
3.2.3 Methylation step
Toluene methylation with DMC and methanol occurs via an identical methylation step.The reaction energy profiles,referencing the DMC or methanol gas phase states,are shown in Fig.3 and Fig.4,respectively.To initiate the methylation step,methoxide/toluene co-adsorption structures are formed via σ-π interaction between the methyl group and the electrons of the aromatic ring of toluene.Ortho-xylene and meta-xylene formation occur from the same co-adsorption species due to the suitable reaction position,whereas para-xylene formation initiates from a different co-adsorbed species.The toluene adsorption energies to a methoxide containing site are-112.9 and-105.0 kJ·mol-1,suggesting a substantial attraction between the methoxide and toluene.
To alkylate,toluene is attacked by the methyl group to form the xylene isomers.Both methylation and deprotonation of toluene occur in a concerted reaction step to both form the xylene product and regenerate the acid site of H-ZSM-5 catalyst.The methylation transition states are denoted as TS_P-Xylene,TS_O-Xylene,and TS_M-Xylene for formation of para-,ortho-,and meta-xylene,respectively.The corresponding structures are illustrated in Fig.5.The geometric parameters of the key species are given in Table 2.In these transition state structures,the C1-O2 bond of methoxide is completely broken and new C1-Cp,C1-Co,and C1-Cmbonds are partially formed.A Ctoluene-Htoluenebond within the toluene molecule is elongated towards deprotonation and reformation of the acid site.The activation energies for the formation of para-,ortho-,and meta-xylene are 63.2,68.2,and 112.9 kJ·mol-1,respectively,when referencing their respective initial co-adsorbed structures.Differences in the rates of the methylation step will dictate selectivity,though with the transition state referenced to the lowest energy toluene co-adsorbed state in all cases,selectivity to para over ortho-xylene is not clearly predicted.
The space restraints of the ZSM-5 pore lead to transition state selectivity against formation of the meta-xylene product,as evidenced by interatomic distances.At the transition state,the attacking methyl group is near to planar,suggesting that much of the positive charge of the reacting species is on this fragment.This methyl fragment is stabilized by interaction with the π electrons of toluene and the negative zeolite framework charge through the O2 atom.The bond distances of C1-Cp,C1-Co,and C1-Cmare 0.222,0.219,and 0.231 nm,respectively.C1-Cmhas the longest interatomic distance.The basic O1 site of the zeolite also assists to stabilize the carbenium ion.The interatomic distances of C1…O1 are both 0.275 nm in TS_P-Xylene and TS_O-Xylene,and the C1…O1 distance is 0.021 nm longer in TS_M-Xylene.This indicates the weakest stabilization effect of the framework of the zeolite on the meta-transition state.Though these distances show little differentiation between forming ortho-or para-xylene,space restraints lead to a greater barrier for meta-xylene production.
Thermodynamically,there is less than a 1.2 kJ·mol?1difference calculated between the three gas-phase products energies.However,there are more significant differences in reaction energies for formation of the adsorbed products.Meta-xylene is the most thermodynamically stable adsorbed product,39.4 and 47.3 kJ·mol-1more stable than adsorbed ortho-xylene and adsorbed para-xylene.The thermodynamic stability is similar between adsorbed para-xylene and ortho-xylene.The desorption energies are 125.9,133.0,172.0 kJ·mol-1for para-,ortho-,and meta-xylene respectively.The desorption energies for xylene desorbing from the acid site inside the pore are higher than that for xylene desorbing from the external surface,which may be due to the additive interaction between xylene and the internal zeolite framework atoms.39At low conversions,selectivity may be presumed to be dictated purely by the kinetic selectivity.At higher conversion,the relative irreversibility of meta-xylene formation could impact selectivity.The desorp-tion process also likely plays an important role in selectively to para-xylene.40The highest desorption energy will cause metaxylene to be retained inside the zeolite and undergo further isomerizationtopara-xylene,increasingthedesiredselectivity.39
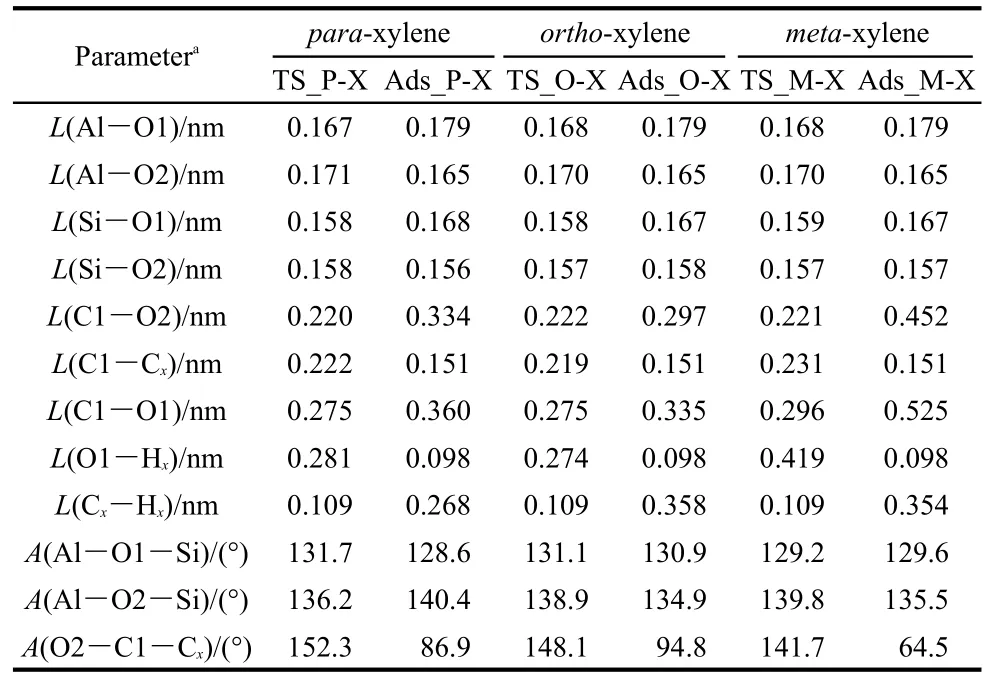
Table 2 Optimized geometric parameters of species involved in toluene methylation with methoxide to form para-,ortho-and meta-xylene over H-ZSM-5
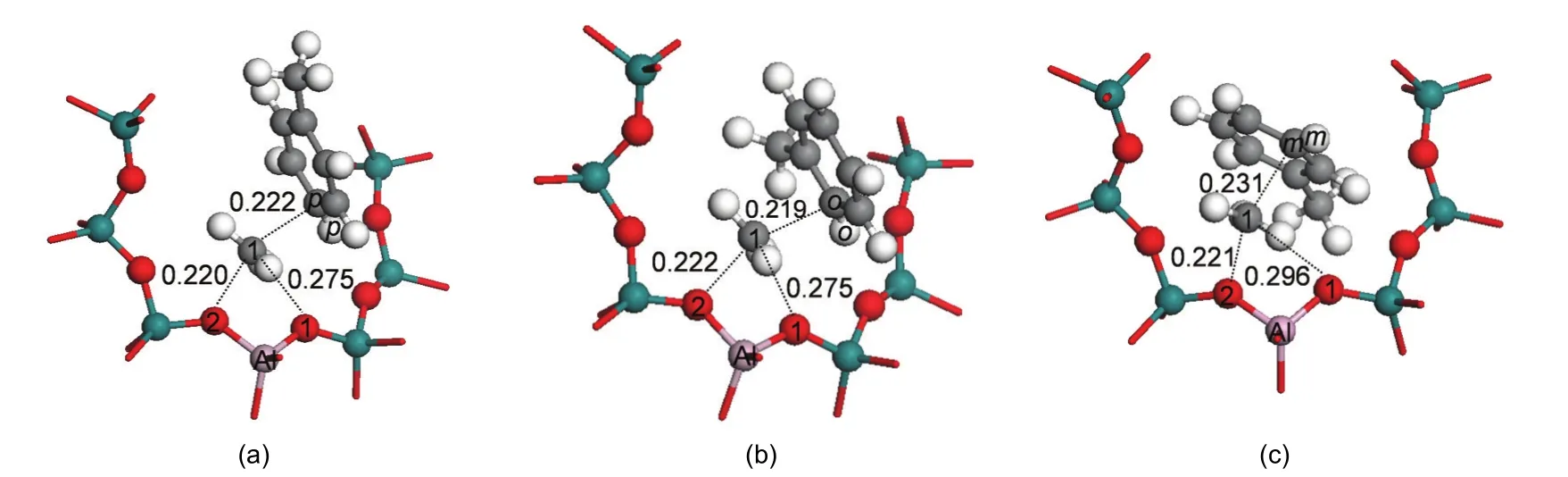
Fig.5 Transition states of toluene methylation with methoxide for formation of(a)para-xylene,TS_P-Xylene;(b)ortho-xylene,TS_O-Xylene and(c)meta-xylene,TS_M-Xylene
Throughout the energetic profiles given in Fig.3 and Fig.4,DMC decomposition and methanol dehydration step are the rate-limiting steps corresponding to toluene stepwise methylation with DMC and methanol,respectively.The DMC alkylation intrinsic activation energies of path A,path B,and path C for the formation of para-,ortho-,and meta-xylene are 186.6,220.5,and 206.7 kJ·mol-1,respectively.Path A of toluene methylation with DMC is kinetically more preferred compared with path B and path C due to the obvious difference in Eavalues.When methanol is the alkylating reagent,the intrinsic activation energy is 120.9 kJ·mol-1for the formation of para-,ortho-,and meta-xylene,which is lower than that of toluene methylation with DMC.For methanol as the alkylating reagent,product desorption energies are larger than the intrinsic activation energy of 120.9 kJ·mol-1,suggesting that product desorption may impact activity and selectivity.The intrinsic activation energy of toluene alkylation with DMC is higher than alkylation with methanol along the methoxide intermediateformation path,which may be attributed to the larger space limiting effect of the zeolite framework to DMC and the more stable hydrogen-bond interaction between methanol and the ZSM-5 framework oxygen atom.
In our considerations of methylation paths,we have not considered possible non-selective decomposition reactions of the methoxide intermediate that could lead to coke build-up.Wang et al.41have suggested that the methoxide species may be deprotonated through hydrogen abstraction to recreate the acid site at the reaction temperatures above 493 K,and that the resulting ylide or carbine species may motivate formation of bulky hydrocarbons leading to coke deposits.Methylation paths that pass through adsorbed methoxide species may be expected to experience a greater amount of coke build-up than paths that avoid this intermediate.In the stepwise path of toluene methylation with methanol,the water molecule could stabilize the methoxide intermediate against these undesired side reactions.Our results suggest that methylation with DMC prefers to occur through a methoxide intermediate,possibly explaining the more rapid deactivation of the H-ZSM-5 catalyst with DMC versus methanol as the alkylating reagent.5
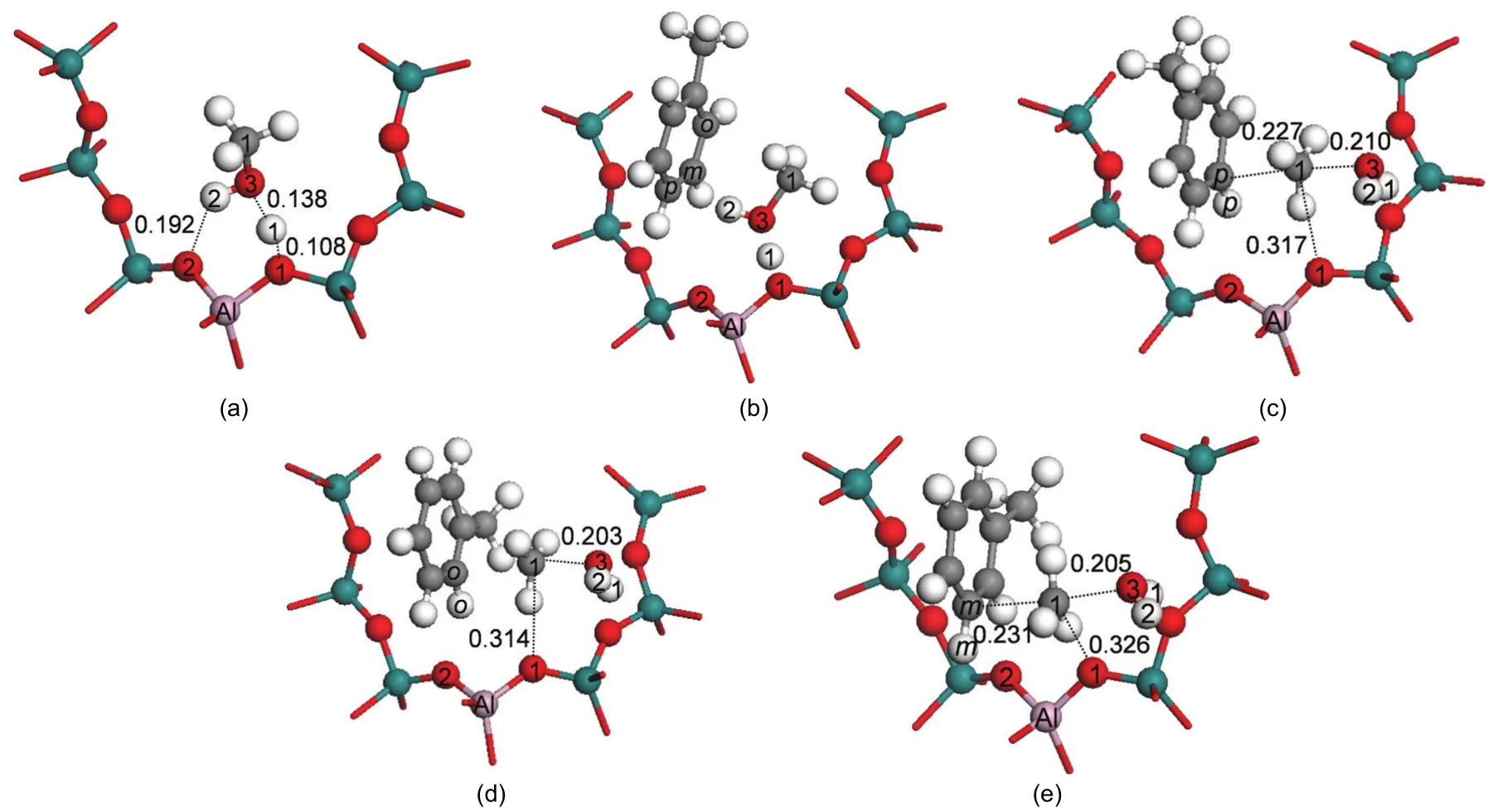
Fig.6 Optimized structures involved in toluene methylation with methanol in the concerted path
3.3 Concerted methylation of toluene with methanol
In addition to the stepwise mechanism,a concerted mechanism may be operable for toluene methylation with methanol(Scheme 1(b)).In the concerted path,a methanol molecule adsorbs over the Br?nsted acid site by two hydrogen bonds forming an end-on adsorption configuration(Ads2_Met)as illustrated in Fig.6(a).11The adsorption energy is-149.8 kJ·mol-1.The toluene molecule co-adsorbs in the vicinity of the methanol molecule,labeled Co_ads and shown in Fig.6(b).The hydrogen bond between the Br?nsted acid(H1)and the O3 atom of methanol remains unchanged.The toluene molecule interacts with methanol by an H-π interaction between the H atom of the methanol hydroxyl group and the aromatic ring of toluene.A similar co-adsorption structure was reported by Mirth and Lercher.14The co-adsorption energy is-231.0 kJ·mol-1.The relative energies of the key states are given in Fig.7.
In the concerted path,methylation to form xylene along with dehydration to form water occurs in a single concerted step catalyzed by H-ZSM-5.The activation energies for this step are 123.4,133.9,and 144.3 kJ·mol-1for formation of para-,ortho-,and meta-xylene.These activation energy barriers are lower than that obtained on a AlSi2O4H9cluster without the stabilizing effect of the H-ZSM-5 pore to the carbenium ion of transition state.42The transition state structures of the concerted dehydration-methylation step are labeled asTS_P-Xylene,TS_O-Xylene,and TS_M-Xylene(shown in Figs.6(c),6(d),and 6(e)),respectively).In the transition states,the C1-O3 bond of methanol is broken,the O3-H1 bond is formed by releasing a water molecule,and the C1-Ctoluene(C1-Cp;C1-Co;C1-Cm)bond is partially formed to produce a carbenium-ion intermediate.As in all transition states reported thus far in this study,a methyl carbenium ion is created in the transition state.The carbenium nature of the methyl group is reflected by the nearly trigonal planar structure.The positive charge in the methyl carbenium is co-stabilized by the O3 atom of the formed water molecule,the π electrons on the aromatic toluene ring,and interaction of the positive charge with the conjugate zeolite base through the O1 site.Key interatomic distances are given in Table 3.In TS_P-Xylene,TS_O-Xylene,and TS_MXylene,the interatomic distances of C1-Cp,C1-Co,and C1-Cmare 0.227,0.232,and 0.231 nm,respectively.The bond distances of C1-O1 are 0.317,0.314,and 0.326 nm,respectively.The greater stability of the para-transition state structure is attributed to the strongest C1-Cpbond of the three transition states,as reflected by the shortest bond distance.The TS_O-Xylene structure is more stable than the TS_M-Xylene structure due to a closer proximity between the carbenium ion and the O1 atom.
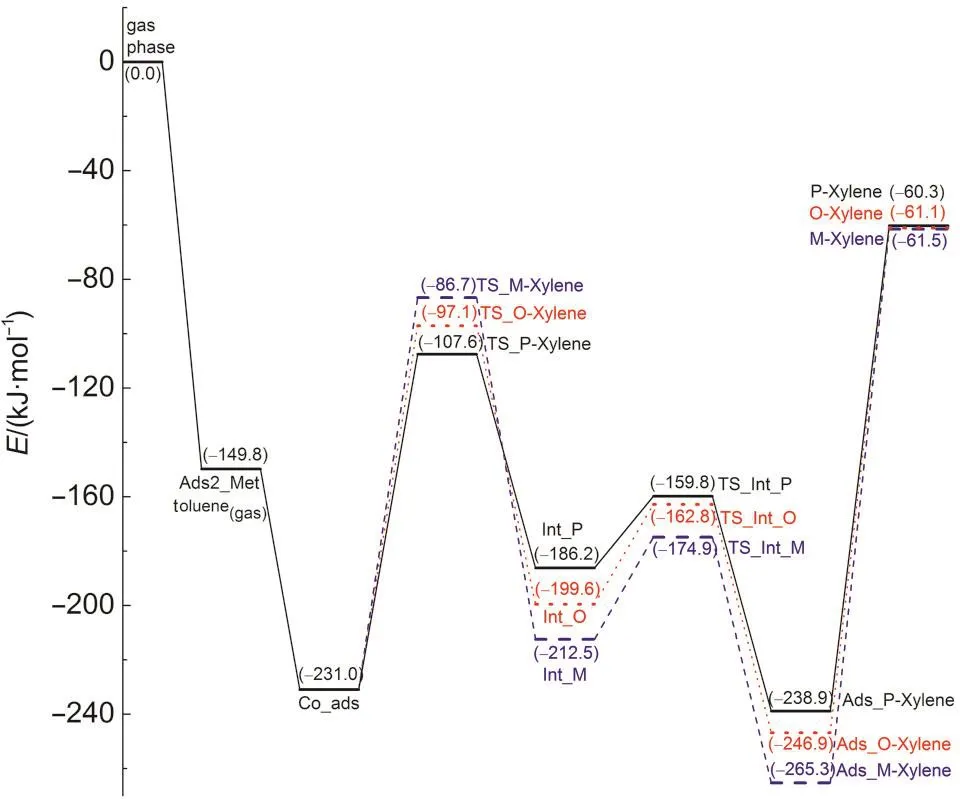
Fig.7 Reaction energy profile for toluene methylation with methanol along the concerted path
Following the methylation step,the generated xylene carbenium ion donates a proton to the O2 site to regenerate the Br?nsted acid site.The Eavalues of the deprotonation steps are 26.4,36.8,and 37.7 kJ·mol-1,respectively,for para-,ortho-,and meta-xylene formation.These values indicate a fast proton-donation,with para-xylene production showing an especially low barrier.The adsorption energy of meta-xylene is 18.4 and 26.4 kJ·mol-1lower than those of ortho-xylene and para-xylene,respectively,which indicates that meta-xylene is the most thermodynamically stable adsorbed product among the isomers.The products diffuse from the acid site via overcoming desorption energies of 178.6,185.8,and 203.8 kJ·mol-1for para-,ortho-and meta-xylene to form gaseous spe-cies and an isolated zeolite.In addition to a lower formation barrier,more rapid desorption may impact the higher selectivity to para-xylene over the other two isomers.
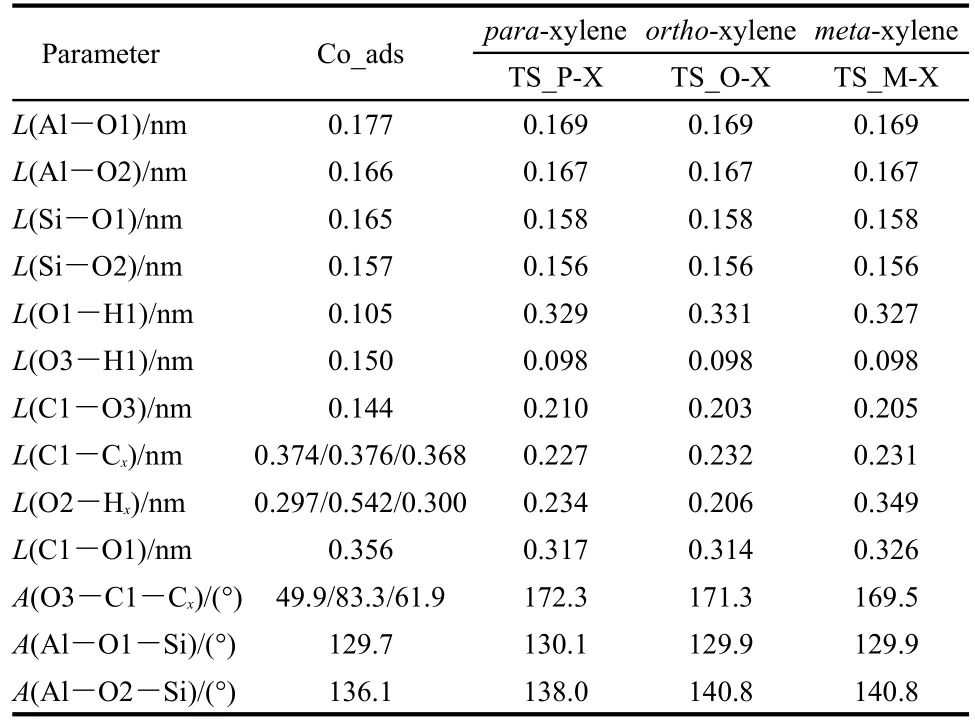
Table 3 Optimized geometric parameters of species involved in toluene concerted methylation with methanol to form the para-,ortho-,and meta-xylene over H-ZSM-5
The intrinsic activation energies of the concerted path for formation of para-,ortho-,and meta-xylene,which is relative to the co-adsorbed methanol and toluene,are dictated by the concerted methylation-dehydration step activation barriers of 123.4,133.9,and 144.3 kJ·mol-1,respectively.The kinetic selectivity order for the formation of xylene is para>ortho>meta in this 128T model,which includes the H-ZSM-5 channel structure.These results agree well with the hard and soft acidity and basicity(HSAB)regulation.12,13
The intrinsic activation energy values we have applied to distinguish the differences between competitive paths cannot be directly used to compare with experimental activation barriers.The experimental barriers of toluene with methanol are referred to the state in which methanol is adsorbed and the aromatic is still in the gas phase,which corresponds to the barriers referencing to the Ads2_Met and toluene in the gas phase state in the concerted path.28The activation barriers referencing this state are termed as“apparent,”to distinguish them from the intrinsic activation energies.28Several experimental studies report apparent activation barriers for toluene methylation with methanol.The apparent activation energy of toluene concerted methylation with methanol is equal to the sum of intrinsic activation energy and the adsorption energy of toluene,which is 42.2,52.7,and 63.1 kJ·mol-1,respectively,for formation paraxylene,ortho-xylene,and meta-xylene.The results are in reasonable agreement with the experimental apparent activation energies estimated by Rabiu and Al-Khattaf43(56.83,67.11,and 79.42 kJ·mol-1for formation para-xylene,ortho-xylene,and meta-xylene,respectively).The average barrier we calculate for formation xylene is 52.7 kJ·mol-1,which is lower than 79.83 kJ·mol-1estimated by Ramakrishna et al.44,but is greater than 47 kJ·mol-1estimated by Odedairo et al.45This average value is in the range of 50-55 kJ·mol-1reported by Vinek and Lercher46.
3.4 Comparison of paths for toluene methylation with methanol
Toluene methylation with methanol catalyzed by H-ZSM-5 can go through either the stepwise path or the concerted path to produce three xylene isomers:para-,ortho-,and meta-xylene.In the stepwise path,the generated water molecule is not involved in the methylation step.The intrinsic activation energy of the methylation step is 120.9 kJ·mol-1for the formation of P-Xylene,O-Xylene,and M-Xylene,respectively.The selectively of meta-xylene is restrained due to the highest Eaof the methylation step and meta-xylene?s higher desorption energy.The stepwise path shows concerted methylation and proton donation back to the initial acid site O1 of H-ZSM-5.In the concerted methylation path of toluene with methanol,the water molecule stabilizes the methylation step.Dehydrogenation to reform the acid site occurs in a second step.The intrinsic activation energy is 123.4,133.9,and 144.3 kJ·mol-1for the formation of para-,ortho-and meta-xylene,respectively.Paraxylene has the highest kinetic selectivity in both the methylation step and the dehydrogenation step.The concerted mechanism shows greater selectivity to para-xylene compared with the stepwise mechanism due to the larger difference in Eavalues.
The intrinsic activation barriers for the stepwise and concerted paths are quite similar.However,the barriers are referred to different initial states,as methanol and toluene are co-adsorbed to start the concerted reaction whereas methanol is adsorbed without toluene in the stepwise path.On the same absolute energy scale,relative to the isolated zeolite,toluene,and methanol molecules,the concerted transition states are more stable than their stepwise counterparts.A more sophisticated kinetic model would be necessary to determine which of the paths is preferred,and whether this varies with the effective pressure of methanol or toluene.
The activation barrier of the kinetically preferred path A of toluene methylation with DMC is 186.6 kJ·mol-1,which is 42.3 kJ·mol-1higher than the highest activation energy of toluene concerted methylation with methanol for formation of meta-xylene.Methanol as the methylation reagent is more active for methylation compared to DMC under otherwise comparable conditions.
3.5 Rate constants of toluene methylation paths
Though a comparison of barriers provides insight into the dominant mechanisms of alkylation for this system,it is further useful to compute rate constants that can allow for more direct comparisons with experiment at reaction temperatures.We therefore computed the rate constants for toluene methylation with DMC and methanol at 773 K.46The rate constant can be estimated as
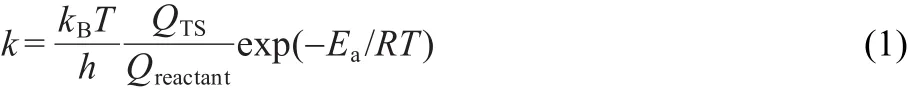
where kBand h are the Boltzmann and Planck constants,R and T are the gas constant and reaction temperature,Q represents the partition function of reactant and transition state,Eadenotes the intrinsic activation energy.
The rate constants,intrinsic activation energies,and the values of QTS/Qreactantare summarized in Table 4.The rate constant data for toluene methylation with DMC illustrate that path A is kinetically preferred compared with paths B and C.Toluene methylation with DMC via path A has a lower rate constant compared with toluene stepwise methylation with methanol due to the higher Ea.The rate constants show that methanol is a more active alkylated reagent than DMC in toluene methylation reaction,in alignment with the lower barriers for alkylation using methanol.For toluene methylation with methanol at 773 K,the stepwise path has a higher rate constant than the concerted path,which is similar with that obtained by Vos et al.42A slightly higher QTS/Qreactantratio in the stepwise path together with the slightly reduced barrier lead to the higher rateconstant for the stepwise path.The difference in barriers and rate constants,however,remains small relative to the accuracy of the DFT methods employed,and without a kinetic model considering the impact of reactant pressures on relative rates,we cannot conclusively determine which of these two paths would be dominant.Kinetic priority of the stepwise path via a methoxide intermediate may affect the stability of the H-ZSM-5 catalyst at high reaction temperature.The highest rate constant of toluene methylation to form para-xylene illustrates that para-xylene is the kinetically preferred product.We note that the conclusions reached with the calculated rate constants are equivalent to those reached by comparing the intrinsic activation barriers,as discussed above.
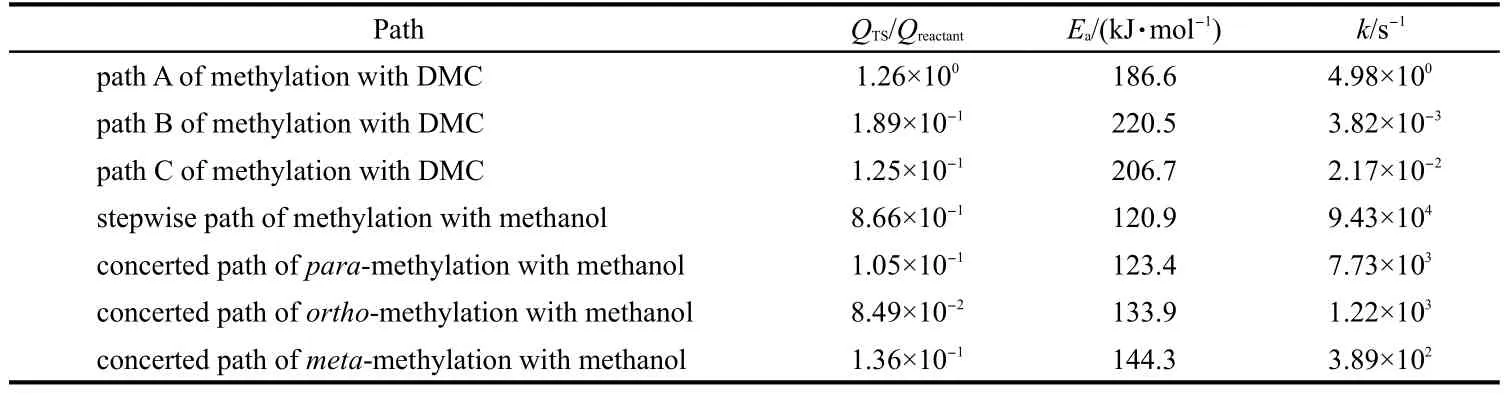
Table 4 Rate constant of toluene methylation with DMC and methanol at 773 K
4 Conclusions
The proposed reaction mechanisms for methylation of toluene with DMC and methanol in an H-ZSM-5 pore have been studied theoretically.Toluene methylation with DMC proceeds through the full decomposition path.Stepwise and concerted paths have similar activation energies for toluene methylation with methanol.The former path has a higher rate constant than the latter path at 773 K.Toluene methylation with DMC needs to overcome a higher activation energy than toluene methylation with methanol.Methanol is more active and economical than DMC in methylation with toluene.For methylation with both reagents,para-xylene formation is kinetically preferred,whereas meta-xylene is the lower energy adsorbed product.The DFT determined reaction energetics provides qualitative and quantitative trends in agreement with reported experimental results.Clarifying that the reaction mechanism is an important step towards enabling further rational design of more active and selective catalysts for this industrially important reaction.
(1)Zhu,Z.R.;Chen,Q.L.;Xie,Z.K.;Yang,W.M.;Li,C.Microporous Mesoporous Mat.2006,88,16.doi:10.1016/j.micromeso.2005.08.021
(2) Inagaki,S.;Kamino,K.;Kikuchi,E.;Matsukata,M.Appl.Catal.A:Gen.2007,318,22.doi:10.1016/j.apcata.2006.10.036
(3)Zhao,Y.;Tan,W.;Wu,H.Y.;Zhang,A.F.;Liu,M.;Li,G.M.;Wang,X.S.;Song,C.S.;Guo,X.W.Catal.Today 2011,160,179.doi:10.1016/j.cattod.2010.05.036
(4)Xue,B.;Li,Y.X.;Deng,L.J.Catal.Commun.2009,10,1609.doi:10.1016/j.catcom.2009.04.028
(5)Tan,W.;Zhao,Y.;Wu,H.Y.;Wang,X.S.;Guo,X.W.Acta Petrolei Sinica 2011,27,719.[譚 偉,趙 巖,吳宏宇,王祥生,郭新聞.石油學報,2011,27,719.]
(6) Liu,S.B.Acta Phys.-Chim.Sin.2009,25,590.[劉述斌.物理化學學報,2009,25,590.]doi:10.3866/PKU.WHXB20090332
(7) Sun,H.;Mumby,S.J.;Maple,J.R.;Hagler,A.T.J.Phys.Chem.1995,99,5873.doi:10.1021/j100016a022
(8) Fu,Y.C.;Zhu,H.Y.;Shen,J.Y.Thermochim.Acta 2005,434,88.doi:10.1016/j.tca.2005.01.021
(9)Kim,W.B.;Kim,Y.G.;Lee,J.S.Appl.Catal.A:Gen.2000,194,403.doi:10.1016/S0926-860X(99)00386-5
(10)Wang,W.;Seiler,M.;Hunger,M.J.Phys.Chem.B 2001,105,12553.doi:10.1021/jp0129784
(11) Ivanova,I.I.;Corma,A.J.Phys.Chem.B 1997,101,547.doi:10.1021/jp961468k
(12) Corma,A.;Llopis,F.;Viruela,P.;Zicovichwilson,C.J.Am.Chem.Soc.1994,116,134.doi:10.1021/ja00080a016
(13) Corma,A.;Sastre,G.;Viruela,P.M.J.Mol.Catal.A:Chem.1995,100,75.doi:10.1016/1381-1169(95)00129-8
(14) Mirth,G.;Lercher,J.A.J.Phys.Chem.1991,95,3736.doi:10.1021/j100162a055
(15)Vos,A.M.;Rozanska,X.;Schoonheydt,R.A.;van Santen,R.A.;Hutschka,F.;Hafner,J.J.Am.Chem.Soc.2001,123,2799.doi:10.1021/ja001981i
(16)Maseras,F.;Morokuma,K.J.Comput.Chem.1995,16,1170.
(17)Dapprich,S.;Komáromi,I.;Byun,K.S.;Morokuma,K.;Frisch,M.J.J.Mol.Struct.-Theochem 1999,462,1.doi:10.1016/S0166-1280(98)00475-8
(18)Nie,X.W.;Janik,J.M.;Guo,X.W.;Song,C.S.J.Phys.Chem.C 2012,116,4071.doi:10.1021/jp209337m
(19) Maihom,T.;Boekfa,B.;Sirijaraensre,J.;Nanok,T.;Probst,M.;Limtrakul,J.J.Phys.Chem.C 2009,113,6654.doi:10.1021/jp809746a
(20)Olson,D.H.;Kokotailo,G.T.;Lawton,S.L.;Meier,W.M.J.Phys.Chem.1981,85,2238.doi:10.1021/j150615a020
(21)Kokotailo,G.T.;Lawton,S.L.;Olson,D.H.;Meier,W.M.Nature 1978,272,437.doi:10.1038/272437a0
(22) Vankoningsveld,H.;Vanbekkum,H.;Jansen,J.C.Acta Crystallogr.,Sect.B Struct.Sci.1987,43,127.doi:10.1107/S0108768187098173
(23) Zhang,J.;Zhou,D.H.;Ni,D.Chin.J.Catal.2008,29,715.[張 佳,周丹紅,倪 丹.催化學報,2008,29,715.]
(24) Li,J.H.;Zhou,D.H.;Ren,J.Acta Phys.-Chim.Sin.2011,27,1393.[李驚鴻,周丹紅,任 玨.物理化學學報,2011,27,1393.]doi:10.3866/PKU.WHXB20110631
(25) Zuo,S.Y.;Zhou,D.H.;Ren,J.;Wang,F.J.Chin.J.Catal.2012,33,1367.[左士穎,周丹紅,任 玨,王鳳嬌.催化學報,2012,33,1367.]
(26)Rappe,A.K.;Upton,T.H.J.Am.Chem.Soc.1992,114,7507.doi:10.1021/ja00045a026
(27) Van Speybroeck,V.;Van der Mynsbrugge,J.;Vandichel,M.;Hemelsoet,K.;Lesthaeghe,D.;Ghysels,A.;Marin,B.G.;Waroquier,M.J.Am.Chem.Soc.2011,133,888.doi:10.1021/ja1073992
(28) Van der Mynsbrugge,J.;Visur,M.;Olsbye,U.;Beato,P.;Bj?rgen,M.;Van Speybroeck,V.;Svelle,S.J.Catal.2012,292,201.doi:10.1016/j.jcat.2012.05.015
(29)Vreven,T.;Morokuma,K.J.Comput.Chem.2000,21,1419.
(30)Chai,J.D.;Head-Gordon,M.Phys.Chem.Chem.Phys.2008,10,6615.doi:10.1039/b810189b
(31) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03,RevisionA.01;Gaussian Inc.:Pittsburgh,PA,2003.
(32) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,RevisionA.02;Gaussian Inc.:Wallingford,CT,2009.
(33) Dibenedetto,A.;Aresta,M.;Giannoccaro,P.;Pastore,C.;Papai,I.;Schubert,G.Eur.J.Inorg.Chem.2006,5,908.
(34) Kirumakki,S.R.;Nagaraju,N.;Chary,K.V.R.;Narayanan,S.J.Catal.2004,221,549.doi:10.1016/j.jcat.2003.09.013
(35)Su,K.;Li,Z.H.;Cheng,B.W.;Liao,K.;Shen,D.X.;Wang,Y.F.J.Mol.Catal.A:Chem.2010,315,60.doi:10.1016/j.molcata.2009.08.027
(36) Aresta,M.;Dibenedetto,A.;Fracchiolla,E.;Giannoccaro,P.;Pastore,C.;Pápai,I.;Schubert,G.J.Org.Chem.2005,70,6177.doi:10.1021/jo050392y
(37)Mazar,M.N.;Al-Hashimi,S.;Bhan,A.;Cococcioni,M.J.Phys.Chem.C 2012,116,19385.doi:10.1021/jp306003e
(38) Lee,C.C.;Gorte,R.J.;Farneth,W.E.J.Phys.Chem.B 1997,101,3811.doi:10.1021/jp970711s
(39)Li,L.L.;Nie,X.W.;Song,C.S.;Guo,X.W.Acta Phys.-Chim.Sin.2013,29,754.[李玲玲,聶小娃,宋春山,郭新聞.物理化學學報,2013,29,754.]doi:10.3866/PKU.WHXB201302063
(40) Zeng,Z.H.,Pan,G.S.Acta Phys.-Chim.Sin.1989,5,145.[曾昭槐,潘貴生.物理化學學報,1989,5,145.]doi:10.3866/PKU.WHXB19890204
(41)Wang,W.;Buchholz,A.;Seiler,M.;Hunger,M.J.Am.Chem.Soc.2003,125,15260.doi:10.1021/ja0304244
(42) Vos,M.A.;Nulens,L.H.K.;Proft,D.F.;Schoonheydt,A.R.;Geerlings,P.J.Phys.Chem.B 2002,106,2026.doi:10.1021/jp014015a
(43) Rabiu,S.;Al-Khattaf,S.Ind.Eng.Chem.Res.2008,47,39.doi:10.1021/ie071038o
(44) Ramakrishna,M.;Subhash,B.;Musti,S.R.Ind.Eng.Chem.Res.1991,30,281.doi:10.1021/ie00050a001
(45) Odedairo,T.;Balasamy,R.J.;Al-Khattaf,S.Ind.Eng.Chem.Res.2011,50,3169.doi:10.1021/ie1018904
(46) Vinek,H.;Lercher,J.J.Mol.Catal.1991,64,23.doi:10.1016/0304-5102(91)85125-L
猜你喜歡
瘋狂英語·新悅讀(2023年4期)2023-09-11 05:37:49
瘋狂英語·新閱版(2023年4期)2023-07-14 10:19:39
故事會(2022年21期)2022-11-06 05:18:38
小學閱讀指南·教研版(2021年5期)2021-09-10 07:22:44
百科探秘·海底世界(2020年8期)2020-07-29 08:57:46
科教導刊(2018年4期)2018-06-08 10:04:22
留學(2018年22期)2018-05-14 13:16:45
短篇小說(2015年4期)2015-12-16 07:00:04
短篇小說(原創(chuàng)版)(2015年4期)2015-05-30 18:40:14
物理與工程(2014年4期)2014-02-27 11:23:09
