
控制CO選擇氧化反應(yīng)中金催化劑熱點形成的新方法
2012-12-12 02:42:56王彩紅李大枝
物理化學(xué)學(xué)報 2012年6期
王 芳 王彩紅 李大枝
(濱州學(xué)院化學(xué)與化工系,山東濱州256603)
1 Introduction
Au supported catalysts have been applied widely on CO oxidation in the absence and presence of H2owing to their attractive catalytic properties.1-8Although factors that affect the performance of these catalysts,such as the Au particle size,the support,and the preparation method,have been studied extensively,studies focused on investigating formation of hot-spot in the process of CO oxidation reaction are relative scarce.9On one hand,a hot region can have a deleterious impact on the reactor performance and may deactivate the catalyst.Moreover, it may lead to severe safety problems by either initiating highly exothermic reactions,or by decreasing the material strength of the reactor wall.Base on the fact that CO and H2oxidations are highly exothermic reactions and the hydrogen oxidation being favored by higher temperautres,an effectively control of temperauture is an essential measure to ensure high CO2selectivity.In general,the appropriate temperature can be obtained by changing the structure of reactors.10-14Several design configurations have been proposed to carry out this process.Although multistaged reactors are able to handle this highly exothermic reaction system with acceptable selectivity,they have the disadvantage of requiring complex hardware to control temperatures,using staged air injections along the catalyst bed.In this paper,we mainly focus on the preparation of supported Au catalysts modified by the deposition-precipitation method.We can find that a FeOxmodified Au/Al2O3catalyst is highly efficient catalyst for CO oxidation in the presence and absence of H2. The temperature increase of catalyst bed can be effectively inhibited by addition of appropriate dopant and optimizing catalyst structure,which results in different product distribution.
2 Experimental
2.1 Catalyst preparation
TheAl2O3-MOx(M=Fe and Zn)composite supports were prepared by the incipient-wetness impregnation method.First,the MOxprecursor of M(NO3)2(≥99.0%)were dissolved in 20 mL distilled water and mixed with calculated amount of γ-Al2O3powder(129 m2·g-1,30-45 mesh).The slurry thus prepared was taken into dryness by continuous stirring and heating (60-70°C).Then,the sample was dried at 120°C overnight, and was subsequently calcined in air at 600°C for 4 h.The goldcontaining catalysts were prepared by the deposition-precipitation method.The adequate amount of HAuCl4·3H2O(Alfa,≥99.99%)was dissolved in 150 mL of deionized water and the pH of the solution was adjusted to 8.0-9.0 by addition of 0.1 mol·L-1NaOH(≥85.0%).The solution was heated to 80°C and then the support was added and kept under continuous stirring for 2 h.The samples obtained were washed several times with deionized water(until the disappearance of Cl-and Na+ions),then dried overnight at 120°C.Fractions from the solids were finally calcined at 300°C for 3 h.The theoretical loading amount of Au is 1.0%(w,mass fraction)and that of MOxis 10%(w).
2.2 Characterizations of catalysts
Before each measurement,the samples were purged with dry air at 300°C for 1 h.Chemical states of Au nanoparticles on the catalysts surface were investigated by X-ray photoelectron spectroscopy(XPS)on a VG ESCALAB 210 Electron Spectrometer(Mg Karadiation;hv=1253.6 eV).XPS data were calibrated using the binding energy of C 1s(285.0 eV)as the standard.The transmission electron microscopy(TEM)images were obtained on a transmission electron microscope (JEM1200-EX,JEOL)with an accelerating voltage of 80 kV.A drop of the solution containing Au nanoparticles was put onto a carbon-supported copper mesh,which was dried at room temperature.High-resolution transmission electron microscopy (HRTEM)images were obtained on a transmission electron microscope(JEM2010,JEOL)with an accelerating voltage of 200 kV.The specific surface area of the catalyst was measured by the Brunauer-Emmett-Teller(BET)method on a Micromeritics ASAP-2010 apparatus at liquid nitrogen temperature with N2as the absorbent at 77 K.
2.3 Activity measurement
Catalytic test was carried out at atmospheric pressure in a fixed bed continuous flow quartz reactor(inner diameter,i.d.8 mm),consisting of a flow controller unit,a reactor unit,and an analysis unit.Typically,100 mg of the catalyst was used in each run.The total flow rate of the feed gas was 60 mL·min-1(gas hourly space velocity(GHSV)=36000 h-1).The feed gas consisted of 5%-20%(φ,volume fraction)of CO and 20%(φ) O2in N2balance.In the process of PROX,a gas mixture containing 55%-25%(φ)H2,2%-5%(φ)CO,and 1%-2%(φ)O2in N2was fed at the flow rate of 30 mL·min-1.Argon was used as the carrier gas and nitrogen was used as the internal standard for gas analysis.The gas phase effluents were analyzed on-line chromatographs equipped with thermal conductivity detector(TCD).At the end of the catalytic tests,the catalyst was cooled under an N2stream and stored for characterizations. The catalytic activities were defined in terms of conversion of CO(ηCO),conversion of O2(ηO2),and selectivity to CO2(S),and were calculated according to the corresponding equations:
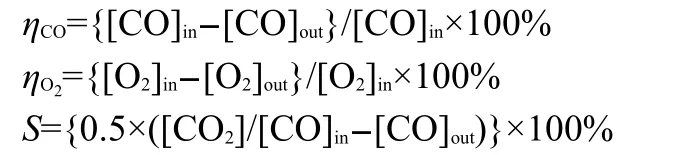
where[CO]inor[O2]inis intake concentration,[CO]outor[O2]outis outtake concentration,[CO2]is production CO2concentration.
3 Results and discussion
3.1 Activity tests
Traditionally,the temperature of catalyst bed measured by thermocouple,as shown in Fig.1(a),could be considered as the reaction temperature.Obviously,it is inaccurate because of external and internal heat transfer hysteresis.We modified the temperature measure system as shown in Fig.1(b),here,thin layer catalyst particles were sandwiched between two inactive quartz sands in tube reactor,the reaction tube was embedded in an adiabatic reactor.In this case,catalyst bed temperature can be obtained directly by thermocouple and its increase completely derives from the exothermic reactions.Although it is difficult to determine the catalyst bed absolutely because it is much dependent on the size,we think it is possible to obtain a relative tendency of the bed temperature behavior qualitatively.
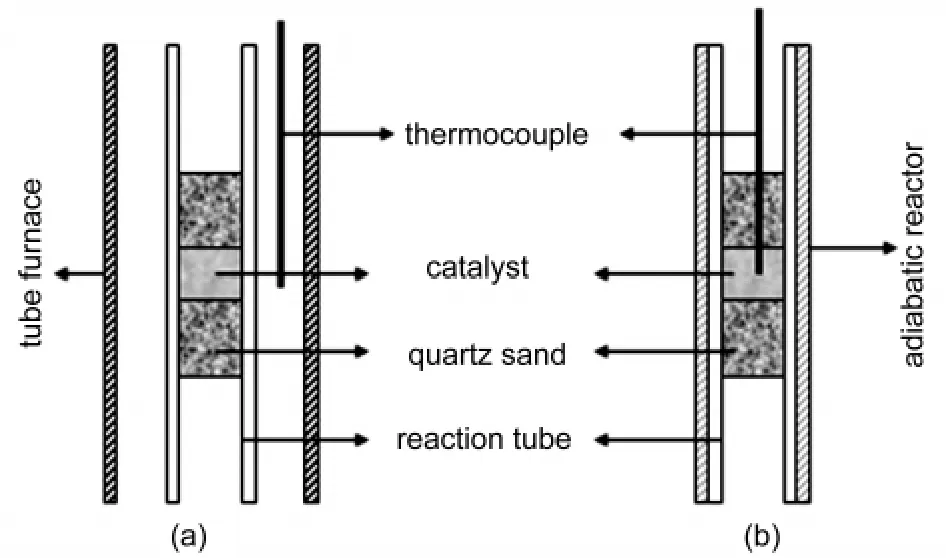
Fig.1 Schematic representation of different reactors
The catalytic activities for CO oxidation in the presence and absence of H2on the original and MOx(M=Fe,Zn)doped Au/ Al2O3catalysts were investigated at ambient temperature.All catalysts exhibited attractive performances for CO oxidation in the absence of H2.CO could be eliminated even when its concentration increased to 20%.However,no measurable activity could be found over Au/Al2O3when H2was introduced in reaction stream.The stabilities of both catalysts dopanted by MOxwere tested in the reaction of CO selective oxidation in the presence of H2and the results were shown in Fig.2.CO could be converted completely and the CO2selectivities over Au/ FeOx-Al2O3and Au/ZnO-Al2O3were 95%and 92%,respectively,even after 100 h.In addition,effects of the volume ratio of O2/CO on CO conversion and CO2selectivity for PROX over Au/FeOx-Al2O3were also measured as shown in Fig.3.It can be found that CO conversion can be stabilized at 100%when the volume ratio of O2/CO increased from 0.5 to 1.0,however, the CO2selectivity decreased from 95%to 50%.
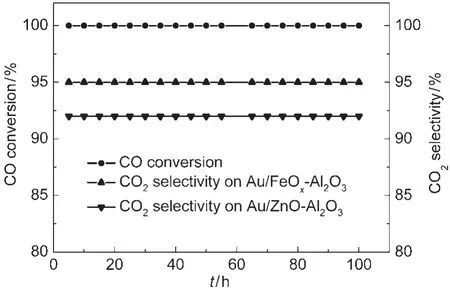
Fig.2 Stability test of theAu/MOx-Al2O3(M=Fe,Zn) catalysts for CO selective oxidation in the presence of H2
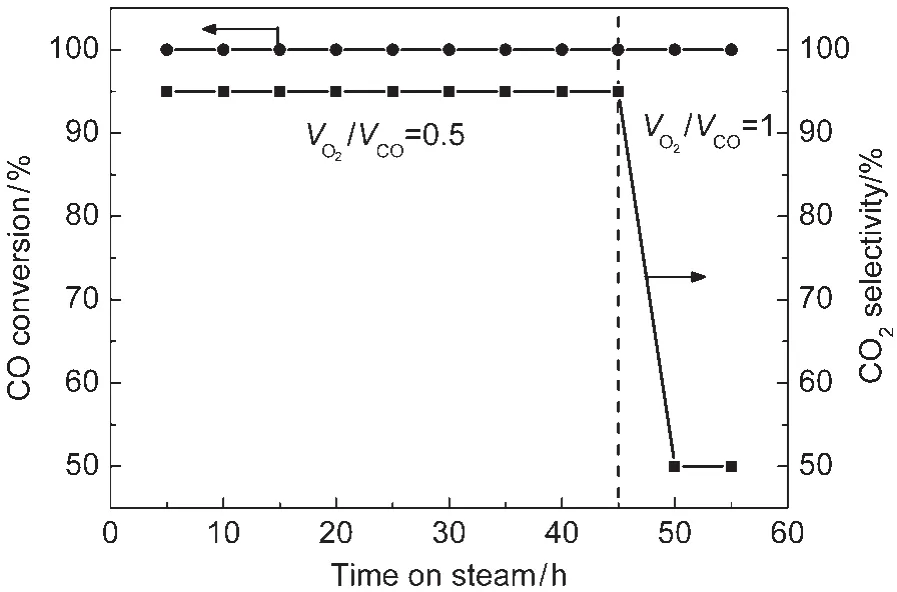
Fig.3 Effects of the volume ratio of O2/CO on CO conversion and CO2selectivity for PROX overAu/FeOx-Al2O3
Based on their attractive catalytic performances,the surface temperatures over various Au catalysts were studied in CO oxidation reaction as shown in Fig.4.All catalyst surface temperatures were enhanced significantly by increasing CO concentration in the reaction stream,owing to reaction exotherm increase.The maximum temperature obtained from Au/Al2O3was 160°C when CO concentration was raised to 20%,whereas the corresponding temperature on the FeOxdoped catalyst was just 55°C.It is interesting to note that the Au/Al2O3catalyst surface temperature could be decreased dramatically by FeOxaddition,especially in higher CO concentration.However,their surface temperatures were enhanced obviously by ZnO addition,a maximum temperature of 170°C could be obtained.The surface temperatures over different Au catalysts in CO selective oxidation in the presence of H2for PROX reaction were also shown in Fig.5.Marwaha et al.15reported that heat generation and heat removal significantly determined the change of product distribution depending on the contact.In order to eliminate effect of contact time,the catalyst surface temperatures were studied in the same space velocity.No detectable temperature changes could be found on the Au/Al2O3catalyst surface because of its low catalytic activity for PROX reaction.However,the surface temperatures on both MOxmodified catalysts could be increased in a certain extent by raising H2concentration in the reaction stream.For the Au/FeOx-Al2O3catalyst,a maximum temperature of 55°C was obtained when CO and H2concentrations were 2.5%and 50%,respectively. However,the surface temperature of the Au/ZnO-Al2O3catalyst could achieve 105°C,and the high temperature resulted in decrease of the corresponding CO2selectivity.The surface temperature transformation of FeOxmodified catalyst was less than 5°C when H2concentrations were increased from 40%to 55%,while CO concentrations were decreased from 4%to 2.5%.These results proved further that the Au/FeOx-Al2O3catalyst is an appropriate candidate for CO oxidation in the absence and presence of H2.Furthermore,the surface temperatures were much dependent on the volume ratio of O2/CO, which might due to that higher ratios were beneficial to the oxidation of hydrogen as shown in Fig.3.
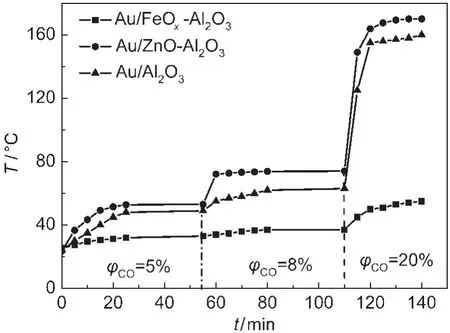
Fig.4 Catalyst surface temperatures in CO oxidation reaction
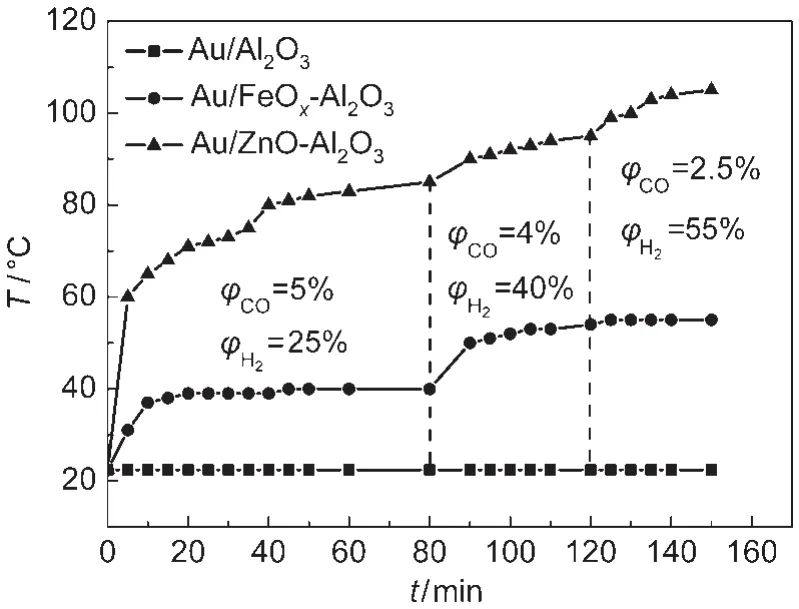
Fig.5 Catalyst surface temperatures in PROX reaction
Five main reactions involved in a PROX reactor include:
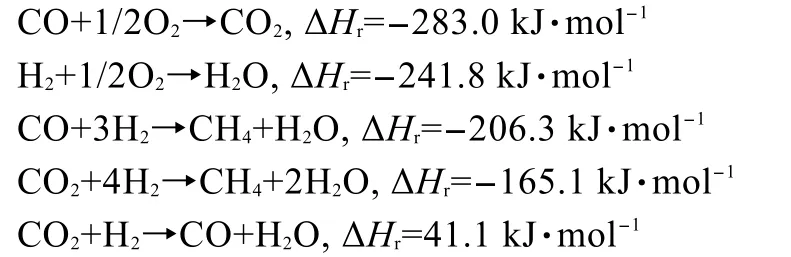
In order to reduce the complexity,only the reactions with significant rates were considered(CO and H2oxidation).The catalyst surface temperature in the adiabatic reactor can be calculated according to the equations16below:
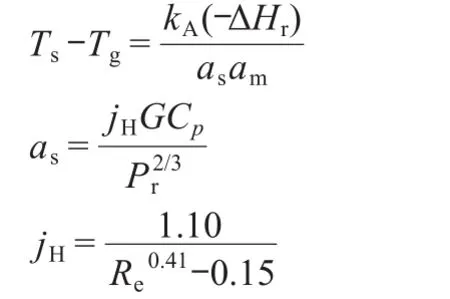
where Tsis catalyst surface temperature,Tgis reaction mixedgas temperature,asis coefficient of heat transfer,amis effective external surface area of catalyst bed per quality unit,and ΔHris exotherm from CO and H2oxidation reactions.Cpis heat capacity of the reaction mixture under constant pressure,and jHis heat transfer factor,while kAand G are reaction rate and mass rate of the reaction mixed-gas.In this case,we consider that the influence of αsand ΔHron Au catalysts surface temperature can be negligeable.It is,however,Tsrather than Tgcontrols the reaction rate kAand also the selectivity of a heterogeneous catalytic process through the Arrhenius equation(K=Aexp(-Ea/ RTs)).17The exponential in Arrhenius?expression for a rate constant has,in fact,not one but two variables,Eaand Ts,which could vary with the imposed experimental conditions and the resulting reaction rates.The traditional interpretation is that the activation energy Eacan be calculated from Tsand the corresponding reaction rates or rate constants.Thus,kAand amare the factors that can affect the catalyst surface temperature.
3.2 Characterization of catalyst
In order to investigate the relationship between various catalyst structures and their surface temperature,a series of characterizations were carried out on various Au catalysts.XPS spectra of Au 4f over the original and MOxdoped Au/Al2O3catalysts are shown in Fig.6.The line shape and width of Au 4f over the original Au/Al2O3catalyst matched well with those of the metallic Au,indicating that zerovalence gold was the active center for CO oxidation at low-temperature.By comparison, the Au 4f features obtained from MOxmodified Au/Al2O3were quite broad,both zerovalence and cationic gold could be found,which wereconsistent with the Au/Fe2O3catalyst reportedpreviously.18In addition,we can find that the AuIII/Au0molar ratio was 53%over Au/ZnO-Al2O3,while that increased to 74%over Au/FeOx-Al2O3catalyst.The result indicated that the valent state of gold particles on the catalyst surface could be changed by MOxaddition.It is already well proven that both AuIIIand Au0species are active for CO oxidation.19Once the catalyst is exposed to a CO/O2mixture,the AuIII/Au0molar ratio decreases and after a sufficiently long exposure only metallic gold is left.This result proves that the lattice oxygen of cationic gold does participate in the reaction of CO oxidation. However,it is should be noted that the decline in activity was not observed when high CO flow rates were employed even for 100 h.The XPS result from Au/FeOx-Al2O3catalyst showed that no AuIII/Au0ratio decline could be found.We consider that cationic gold species does not participate in CO oxidation at low temperature,which maybe play an important role at higher temperature.In other words,the decline in activity caused by the decrease of AuIII/Au0molar ratios can be inhibitted by controlling the hot spot temperature.
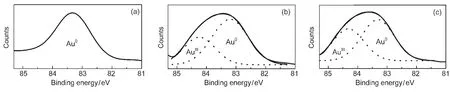
Fig.6 XPS speatra of Au 4f on various catalysts(a)Au/Al2O3,(b)Au/ZnO-Al2O3,(c)Au/FeOx-Al2O3
TEM and HRTEM images from different supported Au catalysts are shown in Fig.7 and Fig.8.The Au nanoparticles on all catalysts had a uniform size around 5 nm and were well dispersed and embedded in mesostructured support matrix.The fringes in Fig.8(a)gave a d-spacing of 0.24 nm,corresponding to the(111)atomic planes of gold lattice.This result shows that the catalysts surface temperature difference can not be attributed to the active component particle size,which is distinguished from NiO/Al2O3catalyst reported previously.20Both polycrystal and single crystal of gold particles coexisted over Au/FeOx-Al2O3,the degree of crystallinity was much lower than that on the Au/Al2O3catalyst.Furthmore,the Au lattice on Au/FeOx-Al2O3preferred to become strongly distorted so as to adopt the lattice dimensions of the mixed-oxide support,which is similar to Au nanoparticles supported on TiO2.21These results indicated that there was a strong interaction between Au and FeOx.Density functional theory(DFT)calculations have shown that lattice strain may enhance surface reactivity.22It has been suggested to explain the unusually high low-temperature CO oxidation activity of small Au particles.In addition,the pore structure parameters of all catalysts from the N2adsorption-desorption isotherms are given in Table 1.The surface area,pore volume,and average pore size of the Au/Al2O3catalyst are 185.0 m2·g-1,0.43 cm3·g-1,and 8.8 nm,respectively.The surface area and average pore size were decreased slightly by ZnO addition.However,it is interesting to note that the surface area of Au/FeOx-Al2O3is smaller than that of the Au/Al2O3catalyst,whereas its average pore size is obviously larger,which maybe due to that FeOxinhibitted part small pores ofAl2O3.

Fig.7 TEM images of different catalysts(a)Au/Al2O3;(b)Au/FeOx-Al2O3;(c)Au/ZnO-Al2O3

Fig.8 HRTEM images of various catalysts(a)Au/Al2O3,(b)Au/FeOx-Al2O3;Inset in Fig.8 shows the corresponding selected area electron diffraction pattern.
Based on the foregoing observations and discussions,we can summarize schematically the adsorption of CO,O2,and/or H2on both catalysts.The difference of the catalyst surface temeratures might mainly due to various active centers,which can result in different reaction mechanisms.With respect to the Au/ Al2O3catalyst,support Al2O3usually can be regarded as chemical inert for CO oxidation.The role of the support material is limited to the stablization of very small gold particles.Thus, oxygen adsorption and dissociation must be possible on the metallic Au nanoparticles.In this case,the activity seems to de-pend very critically on the diameter of the gold particles and only extremely small particles cause highly active samples.No PROX reaction occurs because the active sites are fully covered with H2or CO strongly adsorbed on the Au/Al2O3catalyst due to blocked access of O2to the reaction sites.This strongly indicates that a dissociative adsorption of O2and the following surface reaction with preadsorbed CO are essential for PROX reaction,similarto Langmuir-Hinshelwood mechanism.23While for the Au/FeOx-Al2O3catalyst,on one hand,CO oxidation reaction can occur at the interface between Au and FeOx, just as the Au/Fe2O3catalyst.24Moreover,the addition of FeOxcan enhance the oxygen vacancy,which is in favor of the adsorption of CO and active oxygen species.Therefore,the Au/ FeOx-Al2O3catalyst is very effective for the inhibition of the hot spot formation in CO oxidation reaction,mainly due to the low oxygen affinity of Au,25which results from FeOxmodification.Furthermore,the low temperature of hot spot may be also related to the enhancement of effective external surface area of catalyst bed per quality unit.Herein,we think that there maybe a so-called bi-founctional mechanism existed over the Au/ FeOx-Al2O3catalyst in PROX reaction,just as the Pt-Fe/mordenite catalysts reported previously.26Where Au sites are available for the adsorption of CO as well as H2,and the FeOxsite acts as an O2dissociative-adsoption site.CO adsorbed on a Au site and O2adsorbed on an Fe site react immediately at low temperature once both reactants sit on such neighboring site. The mechanism could not only well explain the excellent catalytic performance of the Au/FeOx-Al2O3catalyst,but also might be responsible for the surface temperature difference between FeOxand ZnO modified catalysts.
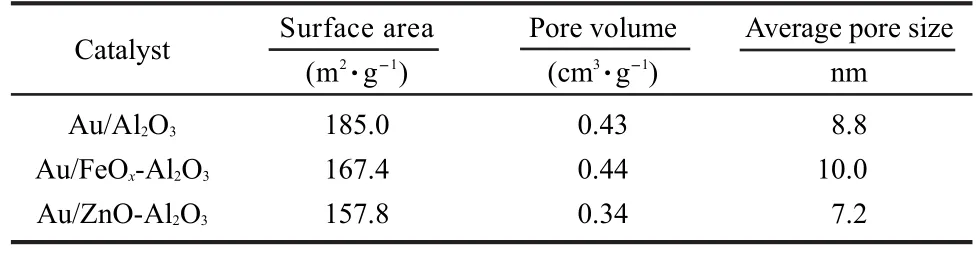
Table 1 Textural properties of different catalysts obtained by N2adsorption
4 Conclusions
In summary,the Au/MOx-Al2O3(M=Zn,Fe)catalysts with high CO oxidation catalytic perfomances in the absence and presence of H2have been synthesized successfully.The temperature increase of catalyst bed can be effectively inhibited by addition of appropriate dopant and optimizing catalyst structure, which results in different product distribution.Reaction path of CO oxidation over alumina supported gold catalyst can be well controlled by the addition of FeOxthrough regulating the oxidation state of Au species.The result opens an alternative line in the investigations on better and more selective catalyst materials.
(1)Yu,J.;Wu,G.S.;Mao,D.S.;Lu,G.Z.Acta Phys.-Chim.Sin. 2008,24,1751.[俞 俊,吳貴升,毛東森,盧冠忠.物理化學(xué)學(xué)報,2008,24,1751.]
(2)Wen,L.;Lin,Z.Y.;Zhou,J.Z.;Gu,P.Y.;Fu,J.K.;Lin,Z.H. Acta Phys.-Chim.Sin.2008,24,581.[文 莉,林種玉,周劍章,古萍英,傅錦坤,林仲華.物理化學(xué)學(xué)報,2008,24,581.]
(3)Ye,Q.;Huo,F.F.;Yan,L.N.;Wang,J.;Cheng,S.Y.;Kang,T. F.Acta Phys.-Chim.Sin.2011,27,2872.[葉 青,霍飛飛,閆立娜,王 娟,程水源,康天放.物理化學(xué)學(xué)報,2011,27,2872.]
(4)Wang,S.R.;Wu,S.H.;Shi,J.;Zheng,X.C.;Huang,W.P.Acta Phys.-Chim.Sin.2004,20,428.[王淑榮,吳世華,石 娟,鄭修成,黃唯平.物理化學(xué)學(xué)報,2004,20,428.]
(5) Liu,Y.L.;You,C.R.;Li,Y.;He,T.;Zhang,X.Q.;Suo,Z.H. Acta Phys.-Chim.Sin.2010,26,2455.[劉玉良,由翠榮,李楊,何 濤,張香芹,索掌懷.物理化學(xué)學(xué)報,2010,26,2455.]
(6) Xu,C.X.;Su,J.X.;Xu,J.H.;Liu,P.P.;Zhao,H.J.;Tian,F.; Ding,Y.J.Am.Chem.Soc.2007,129,42.
(7) Wang,F.;Lu,G.X.Catal.Lett.2007,115,46.
(8) Panzera,G.;Modafferi,V.;Candamano,S.;Donato,A.; Frusteri,F.;Antonucci,P.L.J.Power Sources 2004,135,177.
(9) Zhang,M.H.;Hong,Y.;Ding,S.J.;Hu,J.J.;Fan,Y.X.; Voevodin,A.A.;Su,M.Nanoscale 2010,2,2790.
(10) Kahlich,M.;Gasteiger,H.;Behm,R.J.New Mater. Electrochem.Syst.1998,1,39.
(11) Echigo,M.;Tabata,T.Catal.Today 2004,90,269.
(12) Morillo,A.;Merten,C.;Eigenberger,G.;Hermann,I.;Lemken, D.Chem.Ing.Tech.2003,75,68.
(13) Gritsch,A.;Kolios,G.;Eigenberger,G.Chem.Ing.Tech.2004, 76,722.
(14) Pinkerton,B.;Luss,D.Ind.Eng.Chem.Res.2007,46,1898.
(15) Marwaha,B.;Annamalai,J.;Luss,D.Chem.Eng.Sci.2001,56, 89.
(16) Li,S.F.Chemistry and Catalytic Reaction Engineering; Chemical Industry Press:Beijing,1986;pp 199-202. [李紹芬.化學(xué)與催化反應(yīng)工程.北京:化學(xué)工業(yè)出版社,1986: 199-202.]
(17) Zhu,L.J.;Frens,G.J.Phys.Chem.B 2006,110,18307.
(18) Haruta,M.;Yamada,N.;Kobayash,T.;Iijima,S.J.Catal.1989, 115,301.
(19)Visco,A.M.;Neri,F.;Neri,G.;Donato,A.;Milone,C.; Galvagno,S.Phys.Chem.Chem.Phys.1999,1,2869.
(20) Li,B.T.;Maruyama,K.J.;Nurunnabi,M.;Kunimori,K.; Tomishige,K.Ind.Eng.Chem.Res.2005,44,485.
(21) Graciani,J.;Oviedo,J.;Sanz,J.F.J.Phys.Chem.B 2006,110, 11600.
(22) Mavrikakis,M.;Hammer,B.;N?rskov,J.K.Phys.Rev.Lett. 1998,81,2819.
(23) Morisset,S.;Aguillon,F.;Sizun,M.;Sidis,V.J.Chem.Phys. 2005,122,194702.
(24)Tripathy,A.K.;Kamble,V.S.;Gupta,N.M.J.Catal.1999, 187,332.
(25) Reed,T.B.Free Energy Formation of Binary Compounds;MIT Press:Cambridge,1971.
(26) Kotobuki,M.;Watanabe,A.;Uchida,H.;Yamashita,H.; Watanabe,M.J.Catal.2005,236,262.
猜你喜歡
云南化工(2021年9期)2021-12-21 07:44:16
云南化工(2021年6期)2021-12-21 07:31:42
豬業(yè)科學(xué)(2021年6期)2021-08-12 06:42:16
金橋(2020年11期)2020-12-14 07:52:50
南方農(nóng)業(yè)·中旬(2020年1期)2020-06-21 15:16:40
農(nóng)家科技中旬版(2018年6期)2018-07-31 15:49:38
物理化學(xué)學(xué)報(2018年6期)2018-03-08 03:45:49
農(nóng)民致富之友(2017年5期)2017-04-06 14:00:19
工會信息(2016年1期)2016-04-16 02:38:45
湖北林業(yè)科技(2016年1期)2016-03-25 08:25:42
