
Chemoselective electrochemical seleno-cyclization of dienes to medium-sized benzo[b]azocines
2024-04-05 02:28:38ZhichunWngXinWngQunxinLiShofeiNiDongyngZhoShuinYngGeQiuKiSun
Chinese Chemical Letters 2024年2期
Zhichun Wng ,Xin Wng ,Qunxin Li ,Shofei Ni,* ,Dongyng Zho ,Shuin Yng ,Ge Qiu,c,*,Ki Sun,*
a College of Chemistry and Chemical Engineering, Yantai University, Yantai 264005, China
b Department of Chemistry and Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University,Shantou 515063, China
c Aix Marseille Univ., CNRS, Centrale Marseille, iSm2, Marseille, France
Keywords: Medium-sized N-heterocycles Electrocatalysis Radical Mechanistic insights Selenylation
ABSTRACT The preparation of medium-sized benzo[b]azocines has always been challenging because of inherently unfavorable enthalpy and entropy factors.This report presents a novel approach for accessing 8-membered seleno-benzo[b]azocines via electrochemically-driven seleno-cyclization.This method enables room-temperature preparation of various structurally diverse medium-sized seleno-benzo[b]azocines.The facile deselenation of the seleno-cyclization products to generate functionalized dienes is an additional benefit of this indispensable reaction.Mechanistic insights are presented based on radical inhibition experiments and cyclic voltammetry measurements,which elucidate the radical pathway.Finally,density functional theory calculations further rationalize the rate-determining step and the unique chemoselectivity observed in this transformation.
Medium-sized molecular ring structures,particularly 8-membered benzo[b]azocines,are prominent structural motifs that are found in many natural products and bioactive molecules(Fig.1) [1-8].These ring structures tend to exhibit unique biological activities as analgesic or sedative agents that affect the central nervous system.However,synthesizing 8-membered benzo[b]azocines is difficult,owing to several inherently unfavorable enthalpic and entropic barriers associated with the transition states leading to medium-sized rings [9-14].In recent decades,steady progress has been made in the preparation of mediumsized benzo[b]azocine frameworks,with cycloaddition [15-25] and ring closing metathesis [26-28],representing the most straightforward routes to these 8-memberedN-heterocycles.Nevertheless,most available synthetic procedures require noble transition metal catalysts or unstable starting materials.Therefore,novel strategies should be developed for the facile preparation of medium-sized benzo[b]azocines.

Fig.1.Selected azocinone moieties found in bioactive molecules and natural products.
Organoselenium compounds,especially selenium-containingNheterocycles,are important molecules owing to their roles in medicine and materials science [29-36].Therefore,significant research efforts have been devoted to exploring practical methods for synthesizing selenium-functionalized heterocycles [37].In particular,selenium-centered radical-initiated tandem cyclization at unsaturated bonds represents a valuable strategy for preparing selenium-containingN-heterocycles [38-45].Recently,electrochemical seleno-cyclization across unsaturated bonds [46-50] has demonstrated utility as an environmental-friendly strategy [51,52]for constructing such heterocycles using only electrons to provide the reaction driving force.Moreover,the electrical potential can be precisely controlled during the required electrochemical process,which enables acute manipulation of the chemoselectivity and reactivity [53-56].Building upon our group’s work related to nitrogen heterocyclic chemistry [57-61],herein we designed a diene system comprisingN-(but-3-en-1-yl)-2-vinylanilines and explored electrochemical selenium-centered radical-initiated tandem cyclization.This approach involved unique chemo-and regioselective addition with 8-endo cyclization to obtain a series of 8-membered benzo[b]azocine derivatives.Previous reports have described energetically favorable seleno-radical-based addition cyclizations to selenium-containing 5-or 6-memberedNheterocycles (Scheme 1a) or ionic cycloaddition and ring closing metathesis strategies.However,to our knowledge,this article presents the first successful example of radical-triggered ordered addition involving an 8-endocyclization reaction to prepare medium-sized seleno-benzo[b]azepines (Scheme 1b).

Scheme 1.RSe·-triggered addition cyclization to obtain seleno-heterocycles.
To achieve this goal,critical challenges that originate from the multiple substrate reactive sites and the energetics of cyclization need to be overcome.For example,the diphenylethylene fragment in diene molecules is a powerful radical inhibitor with strong radical trapping capabilities.Therefore,the seleno-radicals generatedinsituare likely to be trapped,thereby terminating the reaction(challenge i).Additionally,in terms of thermodynamics,the secondary carbon radical intermediate might be inclined toward 6-endocyclization to afford an undesirable quinoline scaffold (challenge ii).Finally,considering the two alkenyl groups (activated and unactivated) in the diene system,directing the chemoselective addition of seleno-groups to both double bonds is difficult (challenge iii).
To determine the optimal reaction conditions for the proposed chemo-and regioselective seleno-cyclization,we first carried out the reaction usingN-(but-3-en-1-yl)-4-methyl-N-(2-(1-phenylvinyl)phenyl)benzenesulfonamide1aand diphenyl diselenide2aas model substrates.As shown in Table 1,Pt(+)/Pt(-)were selected as the anode and cathode,respectively,withnBu4NBF4as the supporting electrolyte.The test reaction was performed in 1,2-dichloroethane (DCE) at room temperature under a constant current of 2 mA in an undivided three-necked flask for 3 h,and under these conditions,the target3acould be isolated in 33% yield (entry 1).We then tested other common electrolytes,includingnBu4NI,nBu4NPF6,andnBu4NClO4.The results indicated thatnBu4NPF6exhibited the most positive effect,leading to an isolated yield of3aof 47% (entry 3),whereas reactions withnBu4NI andnBu4NClO4did not proceed efficiently (entries 2 and 4,respectively).Solvent screening revealed thatN,Ndimethylformamide (DMF),tetrahydrofuran (THF),and methanol(MeOH) were not ideal for this transformation (entries 5-7),while dichloromethane (DCM) gave a satisfactory yield of3aup to 60%(entry 8).The impacts of the electrode materials were also explored;however,lower reaction yields were obtained when the Pt(+)/Pt(-) electrodes were replaced with C(+)/C(-) or C(+)/Pt(-)(entries 9 and 10).

Table 1Optimization of reaction conditions.a
Moreover,the yield was not improved appreciably when changing the constant current to 4 mA or appropriately modifying the amount of electrolyte (entries 11-13).A control experiment also confirmed that the desired product3awas not generated without electricity (entry 14).
Next,we used the optimized reaction conditions to examine the substrate scope for the seleno-cyclization of 8-membered seleno-benzo[b]azocines (Scheme 2).Substrates with electronwithdrawing (3-F,4-F,4-Cl,4-Br,5-Br) or electron-donating groups(4-Me,4-OMe,4-p-CNPh,5-Me,5-OMe) on the phenyl ring of the aniline moiety were compatible with this reaction;the transformation proceeded smoothly to afford the corresponding 8-membered seleno-benzo[b]azocines3b-3kin 51%-72% yields.Next,we investigated how substituents on the alkene-tethered phenyl ring influenced the reaction.Substrates bearingo-Me,p-Cl,p-Br,andp-Me groups on the phenyl were all compatible with this conversion,giving the corresponding products3l-3qin 47%-62% yields.When theN-Ts moiety was replaced withN-H,the seleno-cyclization did not occur.
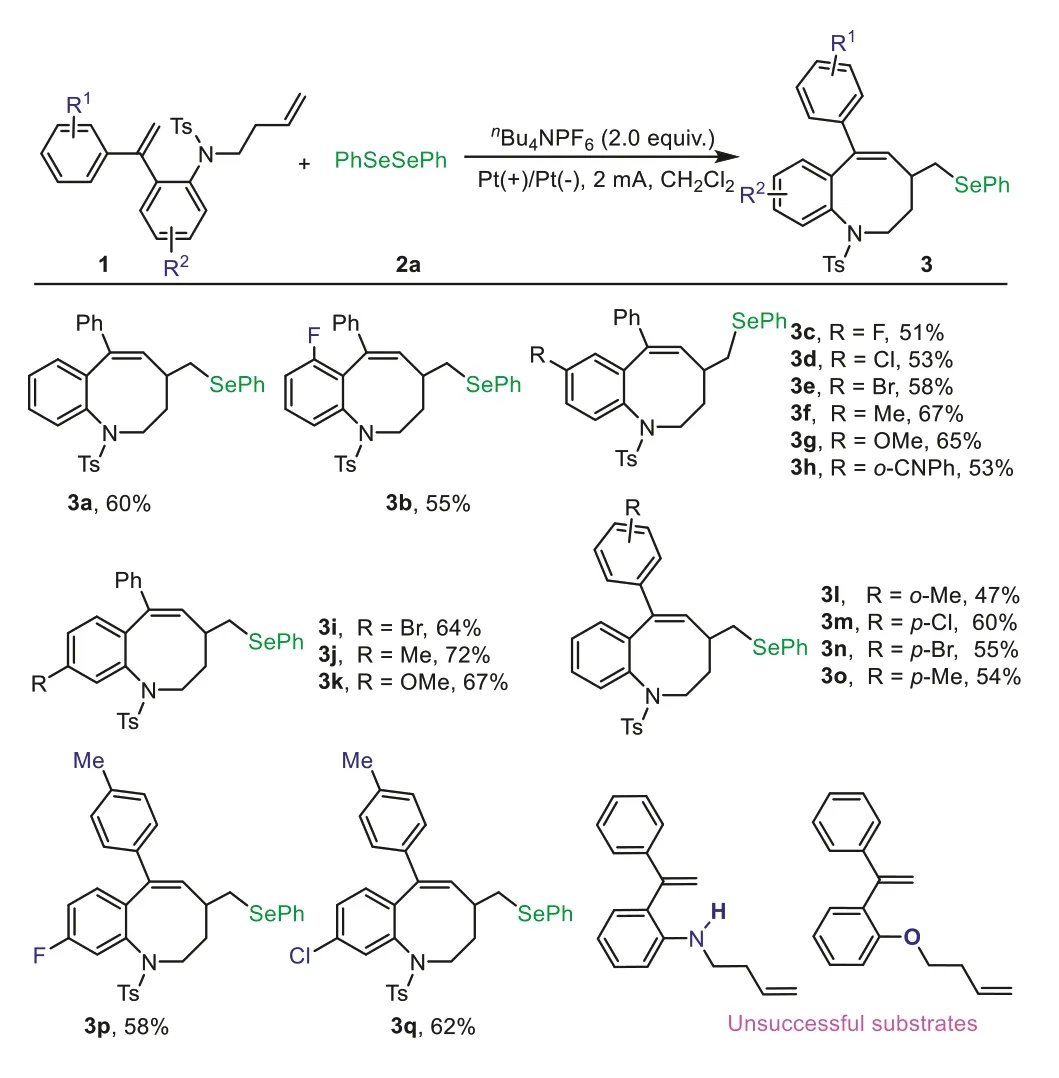
Scheme 2.Diene substrate scope.Reaction conditions: 1 (0.2 mmol),2a (0.2 mmol),supporting electrolyte nBu4NPF6 (0.4 mmol),2 mA constant current,DCM solvent(6 mL),room temperature,12 h,isolated yields are reported.
We further explored the diselenide substrate scope of the seleno-cyclization reaction.As shown in Scheme 3,diselenides bearing electron-donating (-Me,-OMe,-Ph,-tBu) and electronwithdrawing substituents (-Cl,-Br) at various positions on the phenyl ring were all compatible with the reaction,and the corresponding seleno-cyclization products3r-3abwere isolated in moderate to high yields (41%-83%).Steric effects did not appear to impact the seleno-cyclization process because diselenides witho-,m-,andp-Me groups on the phenyl ring proceeded smoothly,and no appreciable yield disparity was observed (3r,3t,and3w).Furthermore,1,2-di(naphthalen-2-yl)diselane and multi-substituted 1,2-bis(3,5-dimethylphenyl)diselane and 1,2-dimesityldiselane were also suitable substrates for this transformation;the corresponding products3ac,3ad,and3aewere obtained in 46%,61%,and 52%yields,respectively.Diselenides with -OH,-NH2,and thiophene substituents were incompatible with this electrochemically-driven transformation.

Scheme 3.Diselenide substrate scope.Reaction conditions: 1a (0.2 mmol),2(0.2 mmol),supporting electrolyte nBu4NPF6 (0.4 mmol),2 mA constant current,DCM solvent (6 mL),room temperature,12 h,isolated yields are reported.
Importantly,the facile conversion of seleno-groups to other functional groups renders selenation reactions indispensable.Treating seleno-benzo[b]azepine3awithm-chloroperbenzoic acid(m-CPBA) or H2O2generated the desired deselenation product4viaoxidation-elimination under mild conditions.This result indicated the potential for further synthetic transformations,such as hydroxylation,oxidation,difunctionalization,and epoxidation(Scheme 4) [62-65].
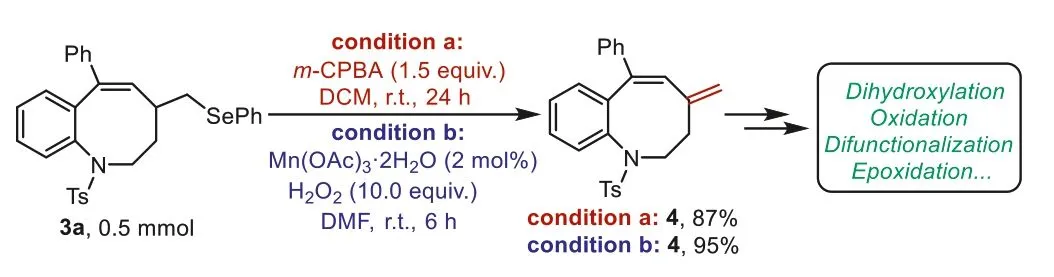
Scheme 4.Deselenation investigations.
Control experiments were performed to gain mechanistic insights regarding this seleno-cyclization reaction (details in Supporting information).First,when a stoichiometric amount of a radical scavenger,e.g.,2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO,2.0 equiv.) or 2,6-di-tert-butyl-4-methylphenol (BHT,2.0 equiv.)was added into the standard reaction system,the selenocyclization was remarkably inhibited,such that substrate1awas recovered (Schemes 5a and b).When stilbene (2.0 equiv.) was added,only 17% of3acould be isolated,and the radical-trapping product5was detectedviahigh resolution mass spectrometry(HRMS) analysis (Scheme 5c).These results suggest that the transformation may proceed through a radical-type mechanism involving a seleno-centered radical.We also conducted a control experiment in a divided cell,and product3awas detected following anodic oxidation (isolated yield=43%;Scheme 5d),indicating that the reaction could be completed directly at the anode.

Scheme 5.Control experiments.
Cyclic voltammetry (CV) experiments were also performed using1aand2a(Fig.2).The oxidation peak of1aappeared at 2.00 V,which was similar to that of2a(1.98 V),consistent with Lei and co-workers’characterization of2a[66].

Fig.2.Cyclic voltammetry results.
On the basis of these experimental results,we proposed a plausible reaction mechanism for the photo-induced 8-endoselenocyclization process (Scheme 6).First,anodic oxidation of (PhSe)22agenerates seleno-radical and seleno-cation intermediates.Steric effects most likely render the seleno-radical more inclined to react with the double bond of the butenyl group to form the favored radical intermediateA,which then undergoes an intramolecular 8-endocyclization to deliver the radical intermediateB.Importantly,the competitive intramolecular 6-endocyclization to a thermodynamically favorable quinoline scaffold (Scheme 1b,challenge ii) was not observed during the transformation.Subsequently,radical intermediateBrapidly undergoes one-electron oxidation at the anode and deprotonation to yield the desired 8-membered benzo[b]azocine3a.Meanwhile,the electrochemical reduction of the DCM solvent or proton occurs at the cathode to balance the overall charge state of the system.

Scheme 6.Proposed mechanism.
Detailed density functional theory (DFT) calculations were performed using the Gaussian 16 package to gain additional mechanistic insights and to determine the origin of the observed chemoselectivity (Fig.3,additional details in Supporting information).The DFT calculations first probed the reaction between theinsitugenerated seleno-radical and substrate1a.As shown in Fig.3a,four potential transition states for the radical addition process could be located depending on the reaction site (i.e.,the double bonds in the butenyl group and diphenylethylene).Radical addition at the terminal carbons of the butenyl double bond (viaTS1-2) and the diphenylethylene (viaTS1-2a) faced lower reaction barriers compared withTS1-2bandTS1-2c.Energy decomposition analysis (Fig.3b) forTS1-2andTS1-2bindicated that the high free energy barrier associated withTS1-2bcould be ascribed to the large geometric distortion from the planer sp2carbon center to the tetrahedralpseudo-sp3carbon center (dihedral angels inTS1-2bandTS1-2measured as 149.6° and 153.7°,respectively).Similar results were obtained from the energy decomposition analysis ofTS1-2aandTS1-2c(Fig.3c),where the planer carbon center was distorted to 138.4° inTS1-2c.We also found thatInt2could isomerize toInt3orInt3’;these two are primed for the subsequent intramolecular cyclization process.These DFT calculations indicated that the free energy barrier required for 8-endocyclizationviaTS3-4was 10.7 kcal/mol,which is 3.5 kcal/mol more favorable than that for the 6-endocyclizationviaTS3-4′.In addition,the 8-endocyclization intermediateInt4is thermodynamically more stable thanInt4’by 18.7 kcal/mol.The DFT calculations also revealed that the cyclization of intermediateInt5toInt6is kinetically and thermodynamically unfavorable.Overall,the computational results support the 8-endocyclization pathway,through whichInt4can be further oxidized and deprotonated to yield the desired 8-membered benzo[b]azocine3a.
In conclusion,we developed a novel method for the electrochemically-driven chemoselective seleno-cyclization of dienes to prepare medium-sized benzo[b]azocines.After compensating for the inherently unfavorable enthalpic and entropic factors and variable chemoselectivity,we obtained 31 structurally diverse seleno-benzo[b]azepines in moderate to high yields at room temperature.The facile deselenation of the seleno-cyclization product to afford new functionalized dienes is an additional valuable feature of this reaction.The inhibition experiments,divided-cell experiments,and CV results were used to propose a plausible radical-based mechanism.DFT calculations further rationalized the rate-determining step and chemoselectivity observed in this transformation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.21801007) and Qingchuang Technology Support Program of University in Shandong Province (No.2021KJ066).S.-F.Ni acknowledges funding from the STU Scientific Research Foundation for Talents (No.NTF20022).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2023.109058.
 Chinese Chemical Letters2024年2期
Chinese Chemical Letters2024年2期
- Chinese Chemical Letters的其它文章
- Hybrid ionic/electronic interphase enabling uniform nucleation and fast diffusion kinetics for stable lithium metal anode
- Multifunctional properties of a polar spin chain compound[N(C3H7)4][Cu(C8H4NO4)]·H2O exhibiting both one-dimensional magnetism and nonlinear optical activity
- Ti3C2Tx MXene wrapped,carbon-coated porous Si sheets for improved lithium storage performance
- Interfacial charge redistribution to promote the catalytic activity of Vs-CoP-CoS2/C n-n heterojunction for oxygen evolution
- Synergy of phosphorus vacancies and build-in electric field into NiCo/NiCoP Mott-Schottky integrated electrode for enhanced water splitting performance
- Ultrathin ternary PtNiGa nanowires for enhanced oxygen reduction reaction