
Emerging targets for glioblastoma stem cell therapy
2016-12-13 08:41:21AhmadSafaMohammadRezaSaadatzadehAaronCohenGadolKarenPollokKhadijehBijangiVishehsaraei
Ahmad R.Safa,Mohammad Reza Saadatzadeh,Aaron A.Cohen-Gadol,Karen E.Pollok, Khadijeh Bijangi-Vishehsaraei
1Indiana University Simon Cancer Center;2Department of Pharmacology and Toxicology;3Department of Neurosurgery,IU School of Medicine and Goodman Campbell Brain and Spine;4Herman B.Wells Center for Pediatric Research,Indiana University School of Medicine,Indianapolis,IN 46202,USA.
Emerging targets for glioblastoma stem cell therapy
Ahmad R.Safa1,2,?,Mohammad Reza Saadatzadeh1,3,Aaron A.Cohen-Gadol3,Karen E.Pollok1,2,4, Khadijeh Bijangi-Vishehsaraei1,2
1Indiana University Simon Cancer Center;2Department of Pharmacology and Toxicology;3Department of Neurosurgery,IU School of Medicine and Goodman Campbell Brain and Spine;4Herman B.Wells Center for Pediatric Research,Indiana University School of Medicine,Indianapolis,IN 46202,USA.
Glioblastoma multiforme(GBM),designated as World Health Organization(WHO)grade IV astrocytoma,is a lethal and therapy-resistant brain cancer comprised of several tumor cell subpopulations,including GBM stem cells (GSCs)which are believed to contribute to tumor recurrence following initial response to therapies.Emerging evidence demonstrates that GBM tumors are initiated from GSCs.The development and use of novel therapies including small molecule inhibitors of specific proteins in signaling pathways that regulate stemness,proliferation and migration of GSCs,immunotherapy,and non-coding microRNAs may provide better means of treating GBM. Identification and characterization of GSC-specific signaling pathways would be necessary to identify specific therapeutic targets which may lead to the development of more efficient therapies selectively targeting GSCs.Several signaling pathways including mTOR,AKT,maternal embryonic leucine zipper kinase(MELK),NOTCH1 and Wnt/β-catenin as well as expression of cancer stem cell markers CD133,CD44,Oct4,Sox2,Nanog,and ALDH1A1 maintain GSC properties.Moreover,the data published in the Cancer Genome Atlas(TCGA)specifically demonstrated the activated PI3K/AKT/mTOR pathway in GBM tumorigenesis.Studying such pathways may help to understand GSC biology and lead to the development of potential therapeutic interventions to render them more sensitive to chemotherapy and radiation therapy.Furthemore,recent demonstration of dedifferentiation of GBM cell lines into CSC-like cells prove that any successful therapeutic agent or combination of drugs for GBM therapy must eliminate not only GSCs,but the differentiated GBM cells and the entire bulk of tumor cells.
glioblastoma multiforme,stem cells,dedifferentiation,CD133,CD44,ALDH1A1,SOX2
Introduction
Glioblastoma multiforme(GBM)is the largest group of brain tumors with very poor response to current therapies[1].Approximately 13,000 people die annually from GBM in the United States,and unfortunately only about 10%of patients survive 5 years[2-4].Despite very significant efforts taken to advance therapeutic strategies for patients with GBM,the clinical prognosis for this devastating disease remains grim.While the combination of radiotherapy and adjunct temozolomide (TMZ)has increased the survival of patients with GBM,the median survival of GBM patients is only about 14.6 months[5].GBM tumors display inter-andintra-patient genomic and phenotypic diversity which originates from the complicated dynamics that form a solid foundation for their development and progression. This genotypic and phenotypic diversity causes variation in tumor response to therapy between patients with the same tumor type.Several factors including the tumor environment,pharmacodynamics,as well as intertumor and intratumoral heterogeneity in individual patients contribute to this variability[6-9].Various genetic alterations which result in redundant and increased cytoprotective and survival pathways as well as numerous defects in the apoptotic signaling machinery and epigenetic alterations contribute to the highly aggressive nature of GBM(Fig.1).The main cause of death in patients with GBM is recurrence of the malignancy which is attributed to treatment-resistant GBM stem cells(GSCs)within the primary tumor and surrounding the tumor.
Substantial evidence indicates that cancer stem cells (CSCs)or cancer-initiating cells(CICs),play a significant role in several cancers,including GBM[10-12].It is well documented that GBM displays a high degree of phenotypic,cellular,genetic,and epigenetic heterogeneity. Furthermore,recent evidence clearly indicates that a major problem in the unresponsiveness of GBM tumors to therapy is the existence of GSCs within the tumor which are most crucial for driving invasive tumor growth and relapse[10,13].Interestingly,in GBM and other malignancies,CSC enrichment may occur either from an increased symmetric self-renewal division rate of CSCs or a reprogramming of non-CSC to CSCs which results in phenotypic plasticity in the tumor population[14-15].The concept of dedifferentiation of non-CSCs to CSCs has increased the complexity of understanding tumor heterogeneity,a potential mechanism for therapeutic relapse,resistance to anticancer therapies,and concerns regarding developing therapeutic strategies.
GBMs harbor dynamic subpopulations of GSCs that have specific phenotypic and genotypic characteristics and can propagate in vivo[15].Since emerging evidence strongly indicates that CSCs are responsible for resistance to therapies in cancerpatients,discovering distinct targets and delineating the molecular differences thatdrive CSC phenotypes hold enormous significance and promise for improving cancer therapy.Particularly, developing targeted inhibitors with the ability to selectively suppress such targets and the molecular drivers of stemness in CSCs may profoundly affect future cancer treatment.In this review article,several signaling pathways that regulate the survival and proliferation of GSCs and the potential of specific proteins in the sepath ways for developing novel and effective inhibitors to eliminate these cells are discussed.Moreover,further understanding GBM cell plasticity and its underlying molecular mechanisms will help in the design of more effective therapies against GBM and preventing tumor recurrence.
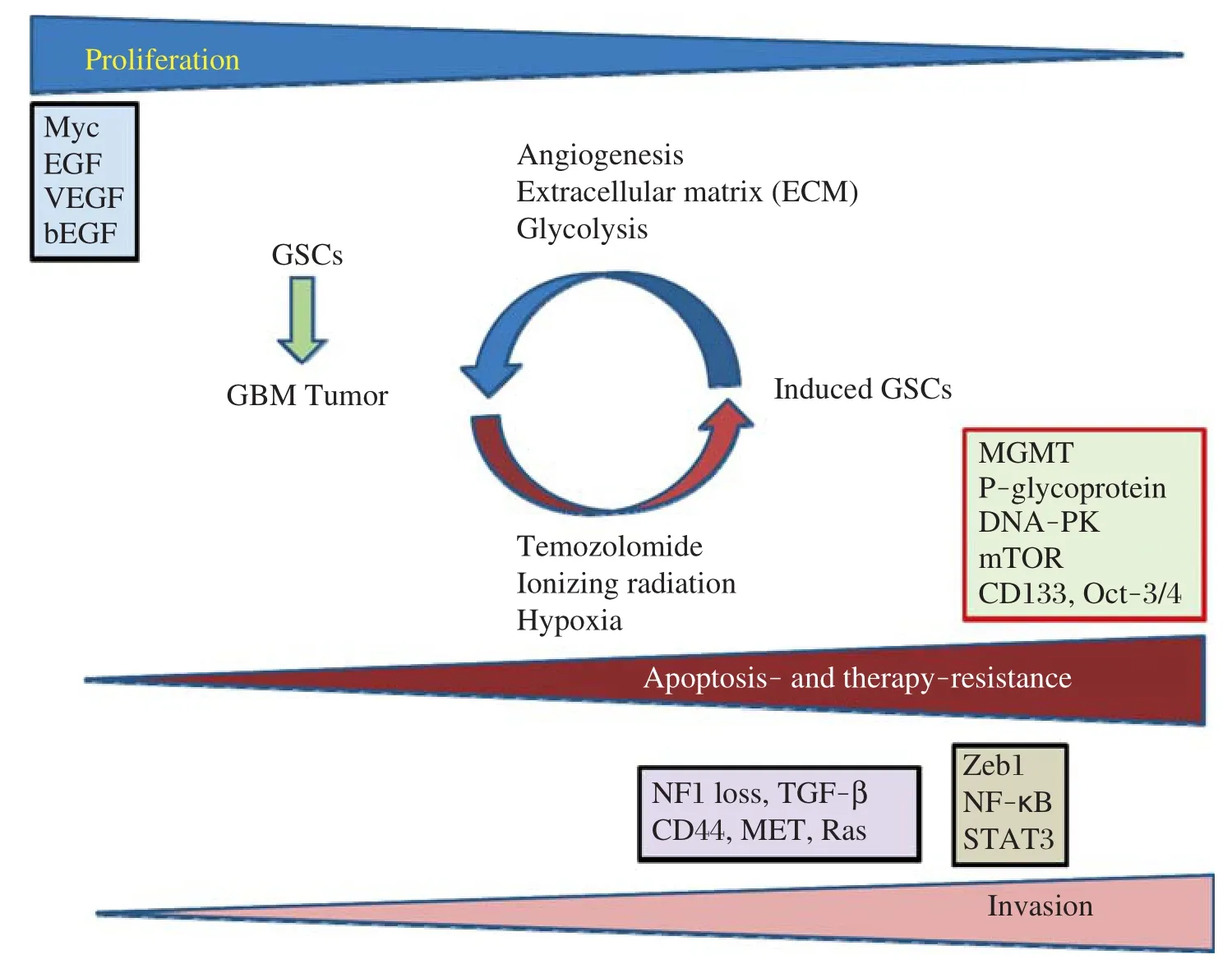
Fig.1Diagram of proliferation,apoptosis-and therapy-resistance,invasion,and the key pathways in glioblastoma multiforme (GBM)and GBM stem cells(GSCs).In this model,the very invasive GBM cells have a slower rate of proliferation,but the highly proliferative cells display a less invasive phenotype.GBM progression and development are determined by three main factors:GBM cell proliferation rate,tumor cell migration,and their resistance to apoptosis and anticancer therapy.Several major proteins including Myc,EGF,VEGF,and bFGF(basic fibroblast growth factor)play major roles in GBM cell proliferation state.Microenvironmental factors(niche)including angiogenesis,tumor extracellular matrix (ECM),and anaerobic glycolysis promote repopulation of tumors and play important roles in regulating the rate of GSC production.Furthermore, chemotherapy,ionizing radiation,and hypoxia can trigger epigenetic plasticity and force induction of GSCs which express proteins involved in apoptosis and therapy resistance including MGMT,P-glycoprotein,DNA-PK,mTOR,CD133,and Oct-3/4.Modified from Xie et al.[96].
GBM
Primary GBM is the most common form of brain tumors[1-2]and is designated as World Health Organization(WHO)grade IV astrocytoma[16-19].Primary GBM is very aggressive and its initiation and recurrence is believed to be caused by GSCs which may be derived from mutated neural stem and precursor cells[13-19]. Furthermore,in contrast to the origin of primary GBM,secondary GBM tumors are developed from lower-grade astrocytomas or oligodendrogliomas. While both types are histologically similar,they are genetically different[15-16].Primary GBM frequently expresses molecular alterations in EGFR,PDGFRA, PTEN,p53,NF1,CDKN2A/B,and telomerase reverse transcriptase(TERT)promoter mutations[21-22]. Furthermore,global hypomethylation is frequently observed in primary human GBM[23].The most detailed information on GBM has been provided by the Cancer Genome Atlas(TCGA)Research Network reporting analysis of copy number,methylation patterns,expression profiling,and whole-genome sequencing of GBM samples[24].A number of genes including EGFR, PDGFRA,CDK4,MDM2,MDM4,MET,CDK6,NMyc,Cyclin D2,PIK3CA,and AKT3 have been found amplified in GBM[20].Additionally,significant abnormalities in several signaling pathways including the receptor tyrosine kinase pathway,the p53 pathway,and the RB pathway were found[17,20,24].Therefore,these data emphasize the complexity in developing therapies to treat GBM.
One molecular marker that is expressed in a subset of GBMcells is the truncated epidermal growth factor receptor(EGFR)mutant referred to as EGFRvIII[25,26].This EGFR variant functions as a ligand-independent constitutively active receptor and displays robust tumorigenic activity and promotes cellular proliferation via activation of the MAPK and phosphatidylinositol-3-kinase(PI3K) Akt pathways[26].This mutation usually occurs in association with the amplification and over expression of wild type EGFR(wtEGFR).However,despite EGFRvIII’s potent ability to enhance tumor igenicity,its expression is usually seen only in a subpopulation of cells[27]. EGFRvIII is an interesting target in GBM therapy because of new EGFRvIII vaccine trials underway[26,28]. Padfield et al.[26]have reviewed the signaling pathways of EGFR and EGFRvIII and the targeted therapy approaches including tyrosine kinase inhibitor,antibody-based therapies,vaccines and pre-clinical RNA-based therapies,as well as the complexities encountered with these molecular targeting approaches including pathway redundancy and intratumoral heterogeneity.
Cancer therapy and drug resistance in CSCs and tumors
In1979,GoldieandColdman[29]proposedthe first simple and elegant mathematical model of drug sensitivity of tumors to their mutation rates.The model indicates that the probability of the appearance of a resistant phenotype increases with the mutation rate.Moreover,for tumors with a nonzero mutation rate the likelihood of there being at least one resistant cell will go from a condition of low to high probability in a very short time.Goldie and Coldman[30]with a simulation approach further expanded their model and explained why an alternating non-crossresistant chemotherapy is optimal.This model stated that the acquisition of multiple levels of drug resistance happens at an accelerated pace and leads to increased degrees of incurability for tumors with a stem cell compartment of a given size.Therefore,based on this model the heterogeneity of slow-growing advanced higher stage clinical tumors will be very high.Furthermore,Foo and Michor[31]reported that drug resistance can emerge due to a host of environmental factors as well as genetic or epigenetic alterations in cancer cells.These factors are particularly important in the interconversion of CSCs to differentiated cells and vice versa[15]which make the therapeutic approaches quite complicated. Notably,evolutionary theory has contributed to our understanding of the dynamics of resistance mutations in a cancer cell population,the risk of resistance preexisting before the initiation of therapy,the drug cocktail composition necessary to prevent the emergence of resistance,and optimum drug administration schedules for patients at risk of evolving acquired resistance[31]. Furthermore,much evidence demonstrates that CSCs contribute to tumor resistance to therapy and recurrence of tumors[15].Adding to the complexity of successful treatment of tumors is the recent demonstration of dedifferentiation of differentiated cancer cells into CSC-likecells due to epigenetic plasticity[15],which proves that any successful therapeutic agent or combination of drugs for cancer therapy must eliminate not only GSCs,but differentiated cancer cells,and the entire bulk of tumor cells.
GSCs signaling pathways
GBM is initiated from the transformation of neural stem cells(NSCs)into GSCs.Similarly,glial progenitors are able to trigger tumor development following malignant transformation of normal progenitor cells[32].Astrocytes,neurons,oligodendrocytes,and ependymal cells also have the potential to initiate tumorigenesis[32].Stem cell-like properties of GSCs could also be acquired during transformation and differentiated nonstem cancer cells can undergo dedifferentiation to form tumor stem-like cells.Recently,Friedmann-Morvinski et al.[33]investigated the mechanisms of dedifferentiation/reprogramming achieved by cortical mature neurons and astrocytes upon transduction with a lentiviral vector containing HRasV12 and shp53.4.These authors confirmed that while transformed dedifferentiated astrocytes and neurons acquired a stem/progenitor cell state,they still retained gene expression memory from their parental cell.Moreover,transcriptional network analysis identified upregulated genes in three main pathways in these cells:Wnt signaling,cell cycle and focal adhesion with thegeneSpp1[also known as osteopontin(OPN)]serving as a common node connecting these pathways.The formation of neurospheres was OPN-dependent,and OPN inhibition in both murine and human glioma tumors prolonged mice survival.These significant results demonstrated that OPN plays an important role in dedifferentiation of cells during tumor formation.Therefore, inhibition of OPN can be a therapeutic target for eliminating GSCs and GBM therapy.
Emerging evidence has demonstrated that GBM originates from CSCs and that GSCs are responsible for cancer patient resistance to therapies[34-36].Therefore, identifying the molecular differences that drive GSC phenotypes and discovering distinct targets will provide significant promise for developing targeted inhibitors for cancer therapy.Fig.2summarizes the characteristics of GSCs which include specific cell surface markers and particular networks of transcription factor(TF)sig-naling,aberrant signaling pathways,epigenetics alterations,reprograming and plasticity,interaction with the microenvironment and GSCs niche,and using particular metabolic pathways.In this review article,several signaling pathways that regulate the survival and proliferation of GSCs and the potential of targeting specific proteins in these pathways to identify and develop novel and effective inhibitors for eliminating these cells are discussed.Characterizing and delineating CSC-specific signaling pathways would help to identify novel therapeutic targets and may lead to the development of more robust and efficient therapies selectively targeting GSCs.Recent work has revealed how normal stem cells (SCs)switch between functional states adjusting to homoeostasis or regeneration[37].This plasticity is also seen in differentiating cells which are capable of reverting to SCs after injury.Similar plasticity in cancer cells has also been reported[15].Interestingly,Olmez et al.[35]recently induced dedifferentiation of patient-derived GBM cell lines into GSC-like cells(induced GBM stem cells,iGSCs)through the expression of Oct4,Sox2 and Nanog transcription factors.Compared with parental GBM cells,iGSCs cells showed significant suppression of EGF receptor and its downstream pathways,formed large neurospheres even in the absence of exogenous mitogens,displayed significant sensitivity to the CSC inhibitor salinomycin,and exhibited resistance to TMZ therapy.Moreover,NOTCH1 and Wnt/β-catenin signaling and expression of CD133,CD44 and ALDH1A1 were induced in iGSCs.These results indicate that dedifferentiation of GBM cells to iGSCs causes complexity in treating this disease and that any therapeutic intervention should be designed to eliminate GSCs as well as iGSCs which may result from treatment with the chemotherapeutic agent TMZ or radiation therapy[15].
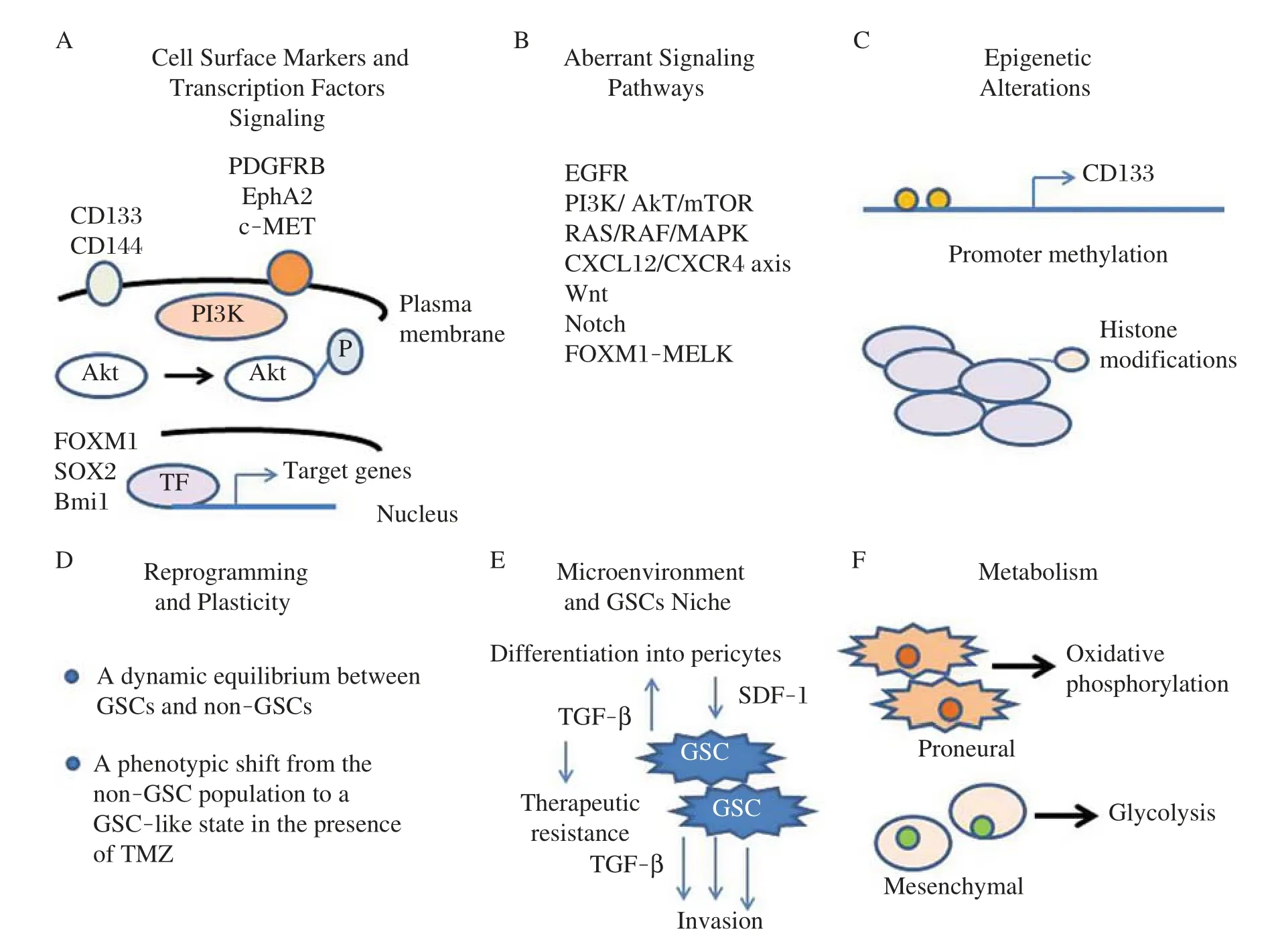
Fig.2Characteristics and potential targets of GSCs.Various factors and cellular processes control GSC phenotype including specific cell surface markers and particular networks of transcription factors(TF)signaling,aberrant signaling pathways,epigenetic alterations,reprograming and plasticity,interaction with the microenvironment and GSCs niche,and particular metabolic pathways.
Studies conducted by the Cancer Genome Atlas (TCGA)also demonstrate the roles of several molecular pathways,specifically the activated PI3K/AKT/mTOR signaling pathway in GSCs and GBM tumorigenesis[20]. Inhibition of multiple molecular pathways may provide an innovative therapeutic approach for managing GBM. Another important protein,the serine/threonine kinase maternal embryonic leucine -zipper kinase(MELK), plays a major role in GSCs.MELK binds and phosphorylates the oncogenic transcription factor FOXM1 in GSCs and recent results show that the catalytic subunit of Polycomb repressive complex2,EZH2,is targeted by the MELK-FOXM1 complex,which in turn promotes GSC resistance to radiation[38].Clinically,EZH2 and MELK are co-expressed in GBM and significantly induced in post-irradiation recurrent tumors whose expression is inversely correlated with patient prognosis[38].Through a gain-and loss-of-function study, these authors show that MELK or FOXM1 contributes to GSC radioresistance by regulating EZH2. Therefore,MELK-FOXM1-EZH2 signaling axis is essential for GSC radioresistance and therefore raises the possibility that MELK-FOXM1-driven EZH2 signaling can serve as a therapeutic target in irradiationresistant GBM tumors.
We were the first to demonstrate the indispensible role of DNA-dependent protein kinase(DNA-PK)in elevating the expression of P-gp and an siRNA or a small molecule inhibitor of DNA-PK abrogating P-gp expression and drug resistance[39].A recent report emphasized the importance of the MDR1 gene and its product P-glycoprotein(P-gp)in GSCs resistance to chemotherapy by showing that following prolonged chemotherapy,DNA-PK,P-gpand CD133+increase in recurrent GBM[40].Interestingly,doxorubicin markedly increased CD133,DNA-PK and MDR1 in GBM cells[40].Their results show that CD133 and DNAPK may increase MDR1 via the PI3K-Akt signal pathway. The PI3K downstream targets Akt and nuclear factor NF-κB,which interacts with the MDR1 promoter,were also increased in these cells.Downregulation of CD133 and DNA-PK by small interfering RNA,or inhibition of PI3K or Akt,decreased Akt,NF-κB and MDR1 expression.Therefore,targeting CD133 and DNA-PK in combination with conventional chemotherapy may effectively eliminate GBM cells and GSCs and improve the prognosis for patients with GBM.
Adherent cultures of GSCs grown on laminin-coated plates(Ad-GSCs)and spheroid cultures of GSCs(Sp-GSCs)expressed high levels of stem cell markers, CD133,Sox2 and Nestin,but low expression of differentiation markers(βIII-tubulin and glial fibrillary acid protein)[41].Recently,Garner et al.[41]have characterized GBM tumors generated by subcutaneous and intracranial injection of Ad-GSCs and Sp-GSCs isolated from a patient-derived xenoline.Interestingly,while these GSCs formed tumors with identical histological features,gene expression analysis showed that xenografts of Sp-GSCs had a classical molecular subtype similar to the bulk tumor cells.In contrast,xenografts generated from Ad-GSCs expressed a mesenchymal gene signature.Furthermore,Ad-GSC-derived xenografts had high STAT3 and ANGPTL4 expression,enriched stem cell markers,transcriptional networks and proangiogenic markers.In clinical samples from GBM patients, STAT3 expression was directly correlated with ANGPTL4 expression.Moreover,increased expression of these genes correlated with poor patient survival. These authors further demonstrated that a pharmacological STAT3 inhibitor suppressed STAT3 binding to the ANGPTL4 promoter and exerted anticancer activity in vivo.Therefore,two distinct sub-populations ofGSCs,Ad-GSCs and Sp-GSCs,generated histologically identical tumors with different gene expression patterns, and a STAT3/ANGPTL4 pathway is only identified in Ad-GSC-derived xeno grafts.Consequently,several targets distinct to each GSC sub-population should be used for more robust GBM therapeutic intervention.
Mao et al.[42]using patient tumor-derived GSCs recently demonstrated that the mitotic E3 ubiquitin ligase CDC20-anaphase- promoting complex(CDC20-APC)drives invasiveness and self-renewal.These authors showed that CDC20 knockdown inhibited and CDC20 overexpression increased the capacity of human GSCs to generate brain tumors in an orthotopic xenograft model in vivo.Moreover,CDC20-APC triggered GSC invasion and self-renewal occurred through pluripotency -related transcription factor SOX2. Therefore,the CDC20-APC/SOX2 signaling axis is indispensible for control of key biological properties of GSCs,and may serve as targeted strategies for the development of novel and effective GBM therapy.
Targeting of CSCs
GBM recurrence occurs largely from remaining CSCs after initial therapy and from epigenetic plasticity and interconversion of the differentiated GBM cells to GSCs resulting from the initial treatment[15,43,44]. Therefore,since the heterogeneity of GSCs increase the complexity of targeting CSCs,any effective and successful GBM therapy must consider eliminating both GSCs as well as the entire bulk of the tumor.Emerging evidence has revealed that the GSC behavior is more dynamic than originally envisioned.Interaction of GSCs with its niche with respect to its exposure to hypoxia and intercellular communication in proximity to endothelial or bone marrow-derived cells(BMDC) may activate GSCs signaling pathways.GBM are exceptionally stroma-rich tumors and may consist of more than 70%stromal components,such as microglia and BMDC. It becomes increasingly apparent that multi-targeted strategies are needed to treat GBM,and recent approaches in GBM therapy include inhibition of invasion(e.g.,integrin,EGFR,CD95,and mTOR inhibition),antiangio genesis and stroma modulators(TGFbeta,VEGF, angiopoetin,and cMET inhibitors),and activation of the immune response(vaccination and blockage of negative co-stimulatory signals)[45].
To eliminate GSCs,one strategy is to use an anti-CSC drug and a drug that epigenetically targets GSCs as well as GBM cells.For example,Booth et al.[46]demonstrated that the lethality of low nanomolar concentrations of salinomycin,a CSC inhibitor,is enhanced by clinically used HDAC inhibitors valproate and vorinostat in GBM cells. These authors demonstrated that regardless of PTEN, ERBB1,or p53 mutational status,salinomycin interacted with HDAC inhibitors in a synergistic fashion to kill GBM cells.The HDAC inhibitor,suberanilohydroxamic acid(SAHA)was recently demonstrated to trigger autophagy through the downregulation of AKT-mTOR signaling,a major suppressive cascade of autophagy[47]. Interestingly,upon pharmacological inhibition of autophagy,SAHA facilitates apoptosis and results in cell death at the early phase,suggesting that SAHA-induced autophagy functions probably act as a prosurvival mechanism. Furthermore,their results also indicated that the inhibition of SAHA-induced autophagy using chloroquine has synergistic effects that further increase apoptosis.These results provide a new perspective on the treatment of GSCs,indicating that SAHA targets GSCs through the induction of autophagy.
Another proposed strategy for targeting GSCs is to first induce differentiation,thus making these cells more amenable to other therapeutic agents.A recent study by Friedman et al.[48]examined this approach by using mTOR inhibition alone and in combination with the differentiating agent all-trans retinoic acid(ATRA)that can target CSCs.The results demonstrated that ATRA, a derivative of retinol,caused differentiation of GSCs as evidenced by the loss of stem cell marker nestin expression.Aberrant function of mTOR has been shown in GSCs.Expression of activated extracellular signalregulated kinase 1/2(pERK1/2)was also increased by ATRA treatment,independent of mTOR pathway inhibitors.The motility of GBM cells was decreased by treatment with ATRA,rapamycin and the PI3K inhibitor LY29002 alone.Interestingly,combination treatment synergistically inhibited effects on GBM cells migration.These findings indicate that ATRA-induced differentiation is mediated via the ERK1/2 pathway and underscores the significance of including differentiating agents along with inhibitors of mTOR pathways in the treatment of GBM.
Combinational inhibition of phosphoinositide-3-kinase/Akt/mammalian target of rapamycin(mTOR) and mitogen-activated protein/extracellular signal-regulated kinase kinase(MEK)/extracellular signal-regulated kinase(ERK)pathways effectively promotes the commitment of glioblastoma cancer stem-like cells(CSLCs)to differentiation and thereby suppresses their tumorigenicity[49].However,the mechanism by which these two signaling pathways are coordinated to regulate differentiation and tumorigenicity remains unknown. FoxO3a,a common phosphorylation target for Akt and ERK,isa major transcription factor which was identified to integrate the signals from the sepath ways[49].Inhibition of Akt and ERK pathways caused nuclear accumulationand activation of FoxO3a more effectively than blockade of either alone,and promoted differentiation of GSCs ina FoxO3a expression-dependent fashion.Furthermore,the expression of aconstitutively active FoxO3a mutant lacking phosphorylation sites for both Akt and ERK was enough to induce differentiation and reduce the tumorigenicity of GSCs.Hence,FoxO3a may play a pivotal role in controlling the differentiation and tumorigenicity of GSCs by the PI3K/Akt/mTOR and MEK/ERK signaling pathways.These data also suggest that developing strategies targeting FoxO3a activation could be potentially useful for the treating GBM.
Di Cristofori[50]recently reported that the vacuolar H+ ATPase(V-ATPase)ATP6V1G1 was unregulated in a series of GBM and GSC neurospheres isolated from GBM patients,and this protein correlated with shorter overall survival.ATP6V1G1 knockdown in GBM neurospheres curtailed sphere-forming ability,induced cell death,and decreased matrix invasion.Treating GSCs neurospheres with bafilomycin A1,the selective V-ATPase inhibitor,reproduced the effects of ATP6V1G1 siRNA and strongly inhibited the expression of the stem cell markers nestin and CD133,and transcription factors SALL2 and POU3F2 in neurospheres.Therefore,ATP6V1G1 serves as a novel marker of poor prognosis in GBM patients and its inhibitors can serve as a new therapeutic strategy for GBM.
A deregulated apoptotic pathway with high levels of the antiapoptotic Bcl-2family of proteins and overt ac tivity of the PI3K signaling pathway has been detected in GBM andGSCs[51].Moreover,ABT-263(Navitoclax),anorally available small-molecule BH-3 mimetic Bcl-2 inhibitor, and GDC-0941,a PI3K inhibitor,abrogated the ability of GSCs to form neurospheres[51].Furthermore,the antiapoptotic Bcl-2 family protein Mcl-1 is overexpressed in GBM and represents an important resistance factor to theBH-3mimeticABT263.GX15-070,a pan-Bcl-2inhibitor which has shown promising antitumor activity in different malignancies,and combined treatment with ABT263 and GX15-070 overcomes apoptotic resistance in established GBM cell lines,glioma stem-like cells, primary cultures,and in in vivo experiments[52].At the molecular level,GX15-070 enhanced apoptosis by posttranslational down-regulation of the deubiquitinase, Usp9X,and the chaperone Bag3,leading to a sustained depletion of Mcl-1 protein levels.Furthermore,knockdown of Usp9X or Bag3depleted endogenous Mcl-1protein levels and in turn enhanced apoptosis induced through Bcl-2/Bcl-xL inhibition[52].Therefore,inhibiting Mcl-1 expression may be a good therapeutic approach and a novel target in GBM therapy.
Micro RNAs and other epigenetic factors in GSCs
Recent results have revealed that microRNAs (miRNAs)play important regulatory roles in the GSC apoptotic pathway,differentiation,proliferation,migration and invasion,drug resistance,and radiation resistance[53-56].Like CSCs from other types of cancer,GSCs are controlled by specific receptor signaling and the regulation of stem cell genes by transcription factors and miRNAs.A number of new targets for these regulators have been identified for GBM treatment and demonstrated that miRNA expression patterns are correlated with the developmental lineage and differentiation state of tumor cells,as well as innovative biomarkers[53-61]. Several published articles have summarized a wide range of miRNAs in GSCs and the molecular mechanisms of miRNAs involved in the signaling pathways regulating these processes,as well as potential usefulness of miRNAs for eliminating GSCs.From the viewpoint of the CSC hypothesis,several deregulated miRNAs have been strongly implicated in regulating the GSCs selfrenewal capacity,maintenance of stemness and plasticity, and resistance to drugs and radiation therapy,as well as unresponsiveness to apoptotic stimuli[57,62-64].Therefore, miRNAs can serve as potential targets for anti-GSC therapeutics[65-69].Using in silico analysis of the Cancer Genome Atlas(TCGA),Wong et al.[55]recently identified miRNAs associated with GBM and GSC survival,and predicted GSC functions in GBM growth and progression.These authors used orthotopic xenograft GBM mouse models and showed that inhibition of miR-148a and miR-31 reduced proliferation,depleted GSCs,normalized tumor vasculature,suppressed tumor growth, and prolonged animal survival.These miRNAs inhibited hypoxia-inducible factor 1(FIH1),and the downstream pathways involving hypoxia-inducible factor HIF1α as well as Notch signaling.Therefore,miR-31 and miR-148a regulate GBM growth by maintaining GSC growth in their niche.
Down-regulation of miRNA-128 may contribute to GBM,in part,by coordinately up-regulating ARP5 (ANGPTL6),Bmi-1 and E2F-3a,resulting in the proliferation of undifferentiated GBM cells[72].A link between miR-128,which is significantly downregulated in GBM,and the loss of GSC self-renewal which occurs by direct regulation of the neural stem cell (NSC)self-renewal factor B lymphoma Mo-MLV insertion region 1 homolog(BMI1)has been shown[73]. The Polycomb Repressor Complex(PRC)is an epigenetic regulator of transcription and its action is mediated by two protein complexes,PRC1 and PRC2.PRC functions as an oncogene in GBM whereit is involved in GSC maintenance and radioresistance. miR-128 directly targets the mRNA of SUZ12,an important component of PRC2,in addition to BMI1, a component of PRC1[74].This reduction of SUZ12 expression blocks the partially redundant functions of PRC1/PRC2,thereby significantly reducing PRC activity and its associated histone modifications.
Epigenetic modifications regulate intratumoral heterogeneity,which is usually regulated by specific GSC niches[76].Moreover,GSC survival,proliferation, and maintenance is regulated by oncogenic cytoprotective signaling pathways and epigenetic modifications (Fig.3)[76].Recently,Nabilsi et al.investigated the extent to which epigenetic differences contribute to intratumoral cellular heterogeneity by developing a high-throughput method,termed MAPit-patch[76]. The authors found several differentially expressed and methylated promoters that are associated with altered gene expression between neural stem cell (NSC)and GBM cell populations.In addition,considering each promoter individually,substantial epigenetic heterogeneity was observed across the sequenced molecules,indicating the presence of epigenetically distinct cellular subpopulations within a GBM tumor[76].Their results showed the biological relevance of epigenetically distinct subpopulations to the phenotypic heterogeneity of tumor cell populations.
Transcriptional mechanisms that control the phenotypic conversion of differentiated tumor cells into tumor-propagating stem-like cells remain to be found. Lopez-Bertoni recently showed that the reprogramming transcription factors Oct4 and Sox2 trigger GBM cells to change into stem-like and tumorpropagating cells via a mechanism involving direct DNA methyltrans ferase(DNMT)promoter transactivation,leading to global DNA methylation and DNMT-dependent downregulation of multiple miRNAs[77].They showed that one of the miRNAs, miRNA-148a,inhibited GBM cell stem-like properties and tumor-propagating potential.These findings identify methylation-and microRNA-based strategies for inhibiting the GSCs,their functions, and contributions to tumor growth and recurrence[77].
Particularly,a significant discovery was recently reported by Kouri et al.[56]who identified miR-182 as a regulator of apoptosis,growth,and differentiationprograms whose expression level is correlated with GBM patient survival[76].The antitumor activity of miR-182 resulted from its repression of Bcl2-like12 (Bcl2L12),c-Met,and hypoxia-inducible factor 2α (HIF2α),as it results in enhanced therapeutic susceptibility,decreased GSC spheroid size,expansion,and stemness in vitro.Intravenously administered 182-SNAs penetrated the blood-brain/blood-tumor barriers (BBB/BTB)in orthotopic GBM xenografts of GBM and selectively disseminated throughout the extravascular glioma parenchyma,triggering reduced tumor burden and increased animal survival.These results represent a novel and powerful strategy for GBM therapy.
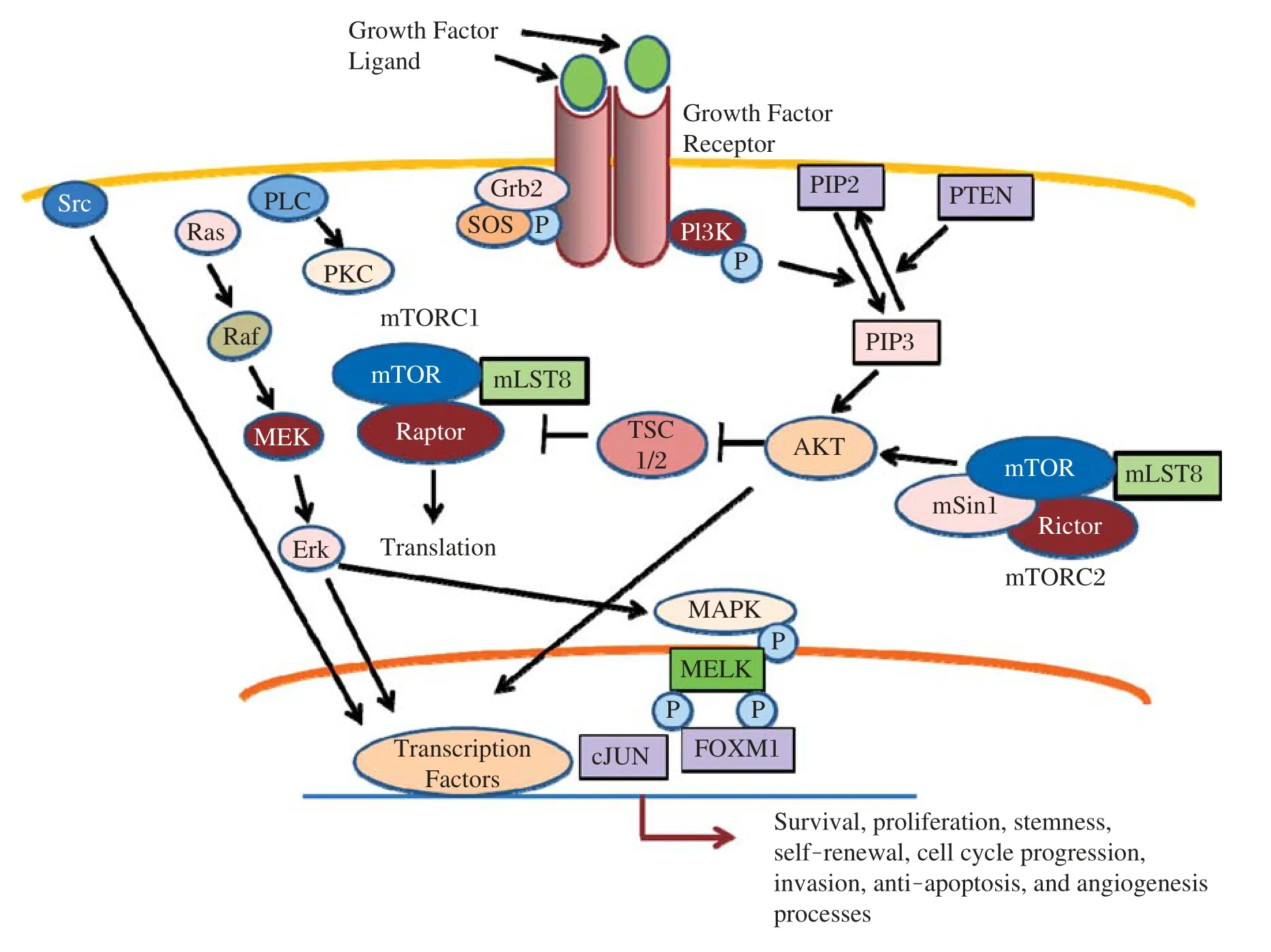
Fig.3Multiple signaling pathways in GSCs.A complex signaling pathway governs self-renewal,stemness,and maintenance of CSGs.Proteins in these pivotal cellular pathways including several plasma membrane receptors,cytoplasmic signaling proteins,specific transcription factors,growth factors,and ligands have the potential to be targeted for eradicating GSCs.
Identification of repurposed drugs for GBM therapy
To discover novel and effective candidate therapeutic drugs for anti-GBM and anti-GSCs,Cheng et al.[78]collected 356 GBM gene signatures from public databases and use the Connectivity Map to systematically evaluate the in vitro antitumor effects of 79 drugs in GBM cell lines and GSCs.Using this strategy,these authors selected the antipsychotic drug thioridazine for further characterization due to its potent anti-GBM and anti-GSC properties.Thioridazine induced autophagy in GBM cells and in vivo as well as suppressed GBM tumorigenesis.This work is particularly important because it provided a new strategy to search for drugs with anticancer and anticancer stem cell properties by using the Connectivity Map and repurposing the antipsychotic drug thioridazine as an effective anti-GBM and anti-GSC agent.
To identify new potent drugs for GBM therapy,Zhouet al.[79]have combined two significant advances in GBM research,(a)brain-penetrating polymeric nanoparticles that can be loaded with drugs and are optimized for intracranial convection-enhanced delivery,and(b)repurposed compounds,previously approved drugs by Food and Drug Administration(FDA),which were identified through library screening to target GSCs.Using fluorescence imaging and positron emission tomography,these authors demonstrated that brain-penetrating nanoparticles can be delivered intracranially in both rats and pigs.Using this strategy,they identified several FDA approved agents that when loaded into brain-penetrating nanoparticles and administered by convection-enhanced delivery,one of these compounds,dithiazanine iodide,significantly increased survival in rats bearing GSC-derived xenografts.
To conceptually improve prognosis in recurrent GBM, a treatment protocol based on a combination of drugs that have not been used as cytotoxic chemotherapy agents but that have been shown to be well tolerated and already marketed and used for other non-cancer diseases was recently developed by Kast et al.[80].These authors found nine drugs and added them to continuous low dose TMZ in patients with recurrent disease after primary treatment with concomitant administration of TMZ with radiotherapy(Stupp Protocol).These drugs were aprepitant,artesunate,auranofin,captopril,copper gluconate,disulfiram, ketoconazole,nelfinavir,and sertraline,and were added to continuous low dose TMZ.The authors discussed each drug and the specific rationale for its use,and how each drug is expected to retard GBM growth and undermine GBM's compensatory mechanisms during TMZ treatment.As discussed in this work,these drug combinations may increase both quality of life and overall survival.
Epigenetic therapy to eradicate GSCs
Two signatures of malignancies including GBM are aberrant gene function and altered patterns of gene expression,and evidence shows that epigenetic changes in collaboration with genetic alterations cause dysregulation in cancer[81-82].The identification and development of drugs to correct aberrant epigenetic changes in CSCs requires knowledge of the extent and roles of epigenetic reprogramming in these cells.The epigenetic changes in cancer are potentially reversible,and treating CSCs with demethylating agents or HDAC inhibitors may potentially reactivate silenced tumor suppressor and TF genes[82].DNA methyltransferase (DNMT)5-azacytidine(Aza)is an effective anticancer agent and inhibitor of GSCs[83-85].The current knowledge of mechanisms underlying the inhibition of DNA methylation by Aza and 5-Aza-2'-deoxycytidine (Aza-dC),and of their apoptotic- and differentiationinducing effects on cancer stem and progenitor cells in various cancers was recently reported[86].Another class of epigenetic inhibitors is HDAC inhibitors. HDACs are a family of proteins that remove acetyl groups from lysine residues of histone proteins and other proteins including TFs[87].These enzymes regulate the conformation and activity of chromatin and mostly function as transcriptional co-repressors as part of large multi-protein complexes[88].Combination of HDAC inhibitors and DNA damaging agents synergistically inhibit growth and induce apoptosis in GSC cells possibly because they promote an open chromatin conformation and allow more effective access of DNA damaging agents to the chromatin,resulting in the increased effectiveness of these agents[17].Recent results have demonstrated that specific combinations of histone methyltransferase and deacetylase inhibitors significantly attenuated GSCs viability but had only a smalleffect on the growth of human bone marrow mesenchymal stem cells(hMSCs)[88].
Conclusions and future directions
While several GSC targeted therapies have been identified,the usefulness of these compounds depends on their pharmacokinetic and toxicity profiles,whether they cross the blood-brain barrier(BBB),and whether they have in vivo activity.A good source of useful agents is repurposing FDA-approved drugs which are clinically used for other diseases and may be effective as single agents or they may have a synergistic effect in combination with TMZ for eliminating the entire bulk of GBM tumors.Among these,drugs that affect epigenetic alterations,including HDAC inhibitors and DNA methyltransferase(DNMT),approved for hematological malignancies,are available for solid tumor therapy[89].Testing against GBM cells and GSCs,several FDA-approved compounds that may be useful in GBM treatment have been identified[90]and led to the rational combination use of statins and topoisomerase inhibitors for GBM therapy[90].Furthermore,Hothi et al.have identified disulfiram(DSF),an FDA approved agent for the treatment of alcoholism,as capable of inhibiting the growth of human GSCs[91].The significance of this finding is that DSF is a relatively nontoxic drug that can cross the BBB,and it is a direct and potent inhibitor of human MGMT in brain tumor cells[91-92].Furthermore,the HDAC inhibitors trichostatin A(TSA)and valproic acid(VPA)are FDA approved drugs that significantly reduced proliferation rates, decreased the expression of cancer stem cell markers, and induced differentiation of these cells[93].These agents may increase the efficacy of conventional cancer treatments for eliminating GSCs.Recent results demonstrated that HDAC inhibitors in combination with erlotinib may be a useful treatment option for newly diagnosed tumors regardless of their EGFR status,as well as for treatment-refractory,EGFR-overexpressing GBM[94].Moreover,the redox agent perylene-quinone hypericin(HYP),a compound targeting multiple epigenetic mechanisms[95],has shown to be effective and GBM patients have displayed stable disease and partial responses to this agent.
While significant information on GSCs has been published,identifying the specific and reliable biomarkers of GSCs is critical for targeting these cells. It is now documented that there are distinct subpopulations of GSCs within a single GBM tumor.Therefore, there is an urgent need to develop agents targeting different signaling pathways and/or employing effective multi-targeting agents to eliminate these distinct GSC subpopulations which display several phenotypic,genotypic and epigenetic characteristics.Several lines of evidence support a model of tumorigenicity with plasticity between the non-GSC and GSC subpopulations within a GBM tumor as well as interconversion of the differentiated non-GSCs to GSCs upon chemotherapy treatment[15].Niche factors that play roles in the interconversion between GSCs and non-GSCs may provide specific niche-related targets to prevent GSC plasticity and dedifferentiation.Moreover, understanding the regulation of interconversion of non-GSCs to GSCs should be high priority research which may potentially lead to the development of rational therapeutic approaches for GBM.
Acknowledgments
We would like to thank Dr.Mary D.Kraeszig for her excellent editorial assistance.This publication was supported in part by the National Cancer Institute of the National Institutes of Health under award number RO1CA138798(KP),the Riley Children's Foundation, the Jeff Gordon Children's Foundation(KP),and the support of the IUPUI Signature Center Initiative for the Cure of Glioblastoma.
References
[1]Jemal A,Murray T,Ward E,et al.Cancer statistics[J].CA Cancer J Clin,2005,55(1):10-30.
[2]Stupp R,Hegi ME,Mason WP,et al.Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study:5-year analysis of the EORTCNCIC trial[J].Lancet Oncol,2009,10(5):459-466.
[3]Smoll NR,Schaller K,Gautschi OP,et al.Long-term survival of patients with glioblastoma multiforme(GBM)[J]. J Clin Neurosci,2013,20(5):670-675.
[4]Hegi ME,Diserens AC,Gorlia T,et al.MGMT gene silencing and benefit from temozolomide in glioblastoma[J]. N Engl J Med,2005,352(10):997-1003.
[5]Stupp R,Mason WP,van den Bent MJ,et al.Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma[J].N Engl J Med,2005,352(10):987-996.
[6]Greaves M,Maley CC.Clonal evolution in cancer[J]. Nature,1212,481(7381):306-313.
[7]Szerlip NJ,Pedraza A,Chakravarty D,et al.Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response[J].Proc Natl Acad Sci USA,2012,109(8):3041-3046.
[8]Snuderl M,Fazlollahi L,Le LP,et al.Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma[J]. Cancer Cell,2011,20(6):810-817.
[9]Sottoriva A,Spiteri I,Piccirillo SG,et al.Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics[J].Proc Natl Acad Sci USA,2013, 110(10):4009-4014.
[10]Singh SK,Hawkins C,Clarke ID.Identification of human brain tumour initiating cells[J].Nature,2004,432(7015):396-401.
[11]Singh AK,Arya RK,Maheshwari S,et al.Tumor heterogeneity and cancer stem cell paradigm:Updates in concept,controversies and clinical relevance[J].Int J Cancer, 2015,136(9):1991-2000.
[12]Yan K1,Yang K,Rich JN.The evolving landscape of glioblastoma stem cells[J].Curr Opin Neurol,2013,26(6):701-707.
[13]Jackson M,Hassiotou F,Nowak A.Glioblastoma stemlike cells:At the root of tumor recurrence and a therapeutic target[J].Carcinogenesis,2015,36(2):177-185.
[14]Gao X,McDonald JT,Naidu M,et al.A proposed quantitative index for assessing the potential contribution of reprogramming to cancer stem cell kinetics[J].Stem Cells Int,2014,2014:249309.
[15]Safa,AR,Saadatzadeh,MR,Cohen-Gadol,AA,et al. Glioblastoma stem cells(GSCs)epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs[J].Genes&Diseases,2015,2(2):152-1637.
[16]Louis DN,Ohgaki H,Wiestler OD,et al.The 2007 WHO classification of tumours of the central nervous system[J]. Acta Neuropathol,2007,114(2):97-109.
[17]Care′n H1,Pollard SM,Beck S.The good,the bad and the ugly:epigenetic mechanisms in glioblastoma[J].Mol Aspects Med,2013,34(4):849-862.
[18]Hamaya K,Doi K,Tanaka T,et al.The determination of glial fibrillary acidic protein for the diagnosis and histogenetic study of central nervous system tumors:a study of 152 cases[J].Acta Med Okayama,1985,39(6):453-462.
[19]Jacque CM,Vinner C,Kujas M,et al.Determination of glial fibrillary acidic protein(GFAP)in human brain tumors[J].Neurol Sci,1978,35(1):147-155.
[20]Masui K,Cloughesy TF,Mischel PS.Review:molecular pathology in adult high-grade gliomas:from molecular diagnostics to target therapies[J].Neuropathol Appl Neurobiol, 2012,38(3):271-291.
[21]Kondo Y,Katsushima K,Ohka F,et al.Epigenetic dysregulation in glioma[J].Cancer Sci,2014,105(4):363-369.
[22]Appin CL,Brat DJ.Molecular genetics of gliomas[J]. Cancer J,2014,20(1):66-72.
[23]Cadieux B1,Ching TT,VandenBerg SR,et al.Genomewide hypomethylation in human glioblastomas associated with specific copy number alteration,methylenetetrahydrofolate reductase allele status,and increased proliferation[J]. Cancer Res,2006,66(17):8469-8476.
[24]Verhaak RG,Hoadley KA,Purdom E,et al.Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA,IDH1, EGFR,and NF1[J].Cancer Cell,2010,17(1):98-110.
[25]Li L,Chakraborty S,Yang CR,et al.An EGFR wild type-EGFRvIII-HB-EGF feed-forward loop regulates the activation of EGFRvIII[J].Oncogene,2014,14(33):4253-4264.
[26]Padfield E,Ellis HP,Kurian KM.Current Therapeutic Advances Targeting EGFR and EGFRvIII in Glioblastoma[J].Front Oncol,2015,5:5.
[27]Inda MM,Bonavia R,Mukasa A,et al.Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma[J].Genes Dev, 2010,24(16):1731-1745.
[28]Johnson LA,Scholler J,Ohkuri T,et al.Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma[J].Sci Transl Med,2015,7(275):275ra22.
[29]GoldieJH,Coldman AJ.A mathematic model for relating the drug sensitivity of tumors to their spontaneous mutation rate[J].Cancer Treat Rep,1979,63(11-12):1727-1733.
[30]Goldie JH,Coldman AJ.Quantitative model for multiple levels of drug resistance in clinical tumors[J].Cancer Treat Rep,1983,67(10):923-931.
[31]Foo J,Michor F.Evolution of acquired resistance to anticancer therapy[J].J Theor Biol,2014,355:10-20.
[32]Goffart N,Kroonen J,Rogister B.Glioblastoma-initiating cells:relationship with neural stem cells and the microenvironment[J].Cancers(Basel),2013,5(3):1049-1071.
[33]Friedmann-Morvinski D,Bhargava V,Gupta S,et al. Identification of therapeutic targets for glioblastoma by network analysis[J].Oncogene 2015 May 11.doi:10.1038/ onc.2015.2119.[Epub ahead of print]
[34]Zhang X,Zhang W,Mao XG,et al.Targeting role of glioma stem cells for glioblastoma multiforme[J].Curr Med Chem,2013,20(15):1974-1984.
[35]Olmez I,Shen W,McDonald H,et al.Dedifferentiation of patient-derived glioblastoma multiforme cell lines results in a cancer stem cell-like state with mitogen-independent growth[J].J Cell Mol Med,2015,19(6):1262-1272.
[36]de Almeida Sassi F,Lunardi Brunetto A,Schwartsmann G,et al.Glioma revisited:from neurogenesis and cancer stem cells to the epigenetic regulation of the niche[J]. J Oncol,2012,2012:537861.
[37]Wabik A,Jones PH.Switching roles:the functionalplasticity of adulttissue stemcells[J].EMBO J,2015,34(9):1164-1179.
[38]Kim SH,Joshi K,Ezhilarasan R,Myers TR,et al.EZH2 protects glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner[J].Stem Cell Reports,2015,4(2):226-238.
[39]Zhong X,Safa AR.Phosphorylation of RNA helicase A by DNA-dependent protein kinase is indispensable for expression of the MDR1 gene product P-glycoprotein in multidrug-resistant human leukemia cells[J].Biochemistry, 2007,46(19):5766-5775.
[40]Xi G,Hayes E,Lewis R,Ichi S,et al.CD133 and DNA-PK regulate MDR1 via the PI3K- or AktNF-κB pathway in multidrug-resistant glioblastoma cells in vitro[J].Oncogene 2015 Mar 30.doi:10.1038/onc.2015.78.[Epub ahead of print]
[41]Garner JM,Ellison DW,Finkelstein D,et al.Molecular heterogeneity in a patient-derived glioblastoma xenoline is regulated by different cancer stem cell populations[J]. PLoS One,2015,10(5):e0125838.
[42]Mao DD,Gujar AD,Mahlokozera T,et al.A CDC20-APC/ SOX2 Signaling Axis Regulates Human Glioblastoma Stem-like Cells[J].Cell Rep,2015,11(11):1809-821.
[43]Natsume A,Ito M,Katsushima K,et al.Chromatin regulator PRC2 is a key regulator of epigenetic plasticity in glioblastoma[J].Cancer Res,2013,73(14):4559-4570.
[44]Jackson M,Hassiotou F,Nowak A.Glioblastoma stemlike cells:at the root of tumor recurrence and a therapeutic target[J].Carcinogenesis,2015,36(2):177-185.
[45]Debus J,Abdollahi A.For the next trick:new discoveries in radiobiology applied to glioblastoma[J].Am Soc Clin Oncol Educ Book,2014;e95-9.
[46]Booth L,Roberts JL,Conley A,et al.HDAC inhibitors enhance the lethality of low dose salinomycin in parental and stem-like GBM cells[J].Cancer Biol Ther,2014, 15(3):305-316.
[47]Chiao MT,Cheng WY,Yang YC,et al.Suberoylanilide hydroxamic acid(SAHA)causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells[J]. Autophagy,2013,10:1509-1526.
[48]Friedman MD,Jeevan DS,Tobias M,et al.Targeting cancer stem cells in glioblastoma multiforme using mTOR inhibitors and the differentiating agent all-trans retinoic acid[J].Oncol Rep,2013,30(4):1645-1650.
[49]Sunayama J,Sato A,Matsuda K,Tachibana K,et al. FoxO3a functions as a key integrator of cellular signals that control glioblastoma stem-like cell differentiation and tumorigenicity[J].Stem Cells,2011,29(9):1327-1337. [50]Di Cristofori A,Ferrero S,Bertolini I,et al.The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma[J]. Oncotarget,2015,6(19):17514-17531.
[51]Pareja F,Macleod D,Shu C,et al.PI3K and Bcl-2 inhibition primes glioblastoma cells to apoptosis through downregulation of Mcl-1 and Phospho-BAD.Mol Cancer Res, 2014;12(7):987-1001.
[52]Karpel-Massler G,Shu C,Chau L,Banu M,et al. Combined inhibition of Bcl-2/Bcl-xL and Usp9X/Bag3 overcomes apoptotic resistance in glioblastoma in vitro and in vivo[J].Oncotarget,2015,6(16):14507-21.
[53]Asuthkar S,Velpula KK,Chetty C,et al.Epigenetic regulation of miRNA-211 by MMP-9 governs glioma cell apoptosis,chemosensitivity and radiosensitivity[J].Oncotarget, 2012,3(11):1439-1454.
[54]Gao X,Jin W.The emerging role of tumor-suppressive microRNA-218 in targeting glioblastoma stemness[J]. Cancer Lett,2014,353(1):25-31.
[55]Wong HK,Fatimy RE,Onodera C,et al.The Cancer Genome Atlas Analysis Predicts MicroRNA for Targeting Cancer Growth and Vascularization in Glioblastoma[J].Mol Ther,2015,23(7):1234-1247.
[56]Kouri FM,Hurley LA,Daniel WL,et al.miR-182 integrates apoptosis,growth,and differentiation programs in glioblastoma[J].Genes Dev,2015,29(7):732-745.
[57]Chu PM,Ma HI,Chen LH,et al.Deregulated microRNAs identified in isolated glioblastoma stem cells:an overview[J].Cell Transplant,2013,22(4):741-753.
[58]Bier A,Giladi N,Kronfeld N,et al.MicroRNA-137 is downregulated in glioblastoma and inhibits the stemness of glioma stem cells by targeting RTVP-1[J].Oncotarget, 2013,4(5):665-676.
[59]Lee HK,Bier A,Cazacu S,et al.MicroRNA-145 is downregulated in glial tumors and regulates glioma cell migration by targeting connective tissue growth factor[J].PLoS One,2013,8(2):e54652.
[60]Yao Y,Ma J,Xue Y,et al.MiR-449a exerts tumor-suppressive functions in human glioblastoma by targeting Myc-associated zinc-finger protein[J].Mol Oncol,2015; 9(3):640-656.
[61]Fazi F,Blandino G.MicroRNAs:non coding pleiotropic factors in development,cancer prevention and treatment[J].Microrna,2013,2(2):81.
[62]Ma J,Yao Y,Wang P,et al.MiR-152 functions as a tumor suppressor in glioblastoma stem cells by targeting Kru¨ppel-like factor 4[J].Cancer Lett,2014,355(1):85-95.
[63]Shang C,Guo Y,Hong Y,et al.MiR-21 up-regulation mediates glioblastoma cancer stem cells apoptosis and proliferation by targeting FASLG[J].Mol Biol Res,2015, 42(3):721-727.
[64]Rathod SS,Rani SB,Khan M,et al.Tumor suppressive miRNA-34a suppresses cell proliferation and tumor growth of glioma stem cells by targeting Akt and Wnt signaling pathways[J].FEBS Open Bio,2014,4:485-495.
[65]Gonza′lez-Go′mez P,Sa′nchez P,et al.MicroRNAs as regulators of neural stem cell-related pathways in glioblastoma multiforme[J].Mol Neurobiol,2011,44(3):235-249.
[66]Yang CM,Chiba T,Brill B,et al.Expression of the miR-302/367 cluster in glioblastoma cells suppresses tumorigenic gene expression patterns and abolishes transformation related phenotypes[J].Int J Cancer,2015 May 20.doi:10.1002/ijc.29606.[Epub ahead of print]
[67]LiuJ,Albrecht AM,NiX,etal.Glioblastoma tumor initiating cells:therapeutic strategies targeting apoptosis and micro RNA pathways[J].Curr Mol Med,2013,13(3):352-357.
[68]Liu Q,Nguyen DH,Dong Q,et al.Molecular properties of CD133+glioblastoma stem cells derived from treatment-refractory recurrent brain tumors[J].J Neurooncol, 2009,94(1):1-19.
[69]Ruan J,Lou S,Dai Q,et al.Tumor suppressor miR-181c attenuates proliferation,invasion,and self-renewal abilities in glioblastoma[J].Neuroreport,2015,26(2):66-73.
[70]Tezcan G,Tunca B,Bekar A,et al.Micro RNA expression pattern modulates temozolomide response in GBM tumors with cancer stem cells[J].Cell Mol Neurobiol,2014,34(5):679-692.
[71]Tezcan G,Tunca B,Bekar A,et al.Olea europaea leaf extract improves the treatment response of GBM stem cells by modulating miRNA expression[J].Am J Cancer Res,2014,4(5):572-590.
[72]Cui JG,Zhao Y,Sethi P,et al.Micro-RNA-128(miRNA-128)down-regulation in glioblastoma targets ARP5 (ANGPTL6),Bmi-1 and E2F-3a,key regulators of brain cell proliferation[J].J Neurooncol,2010,98(3):297-304.
[73]Godlewski J,Nowicki MO,Bronisz A,et al.Targeting of the Bmi-1 oncogene/stem cell renewal factor by micro RNA-128 inhibits glioma proliferation and selfrenewal[J].Cancer Res,2008,68(22):9125-9130.
[74]Peruzzi P,Bronisz A,Nowicki MO,et al.Micro RNA-128 coordinately targets Polycomb Repressor Complexes in glioma stem cells[J].Neuro Oncol,2013,15(9):1212-1224.
[75]Schonberg DL,Lubelski D,Miller TE,et al.Brain tumor stem cells:Molecular characteristics and their impact on therapy[J].Mol Aspects Med,2014,39:82-101.
[76]Nabilsi NH,Deleyrolle LP,Darst RP,et al.Multiplex mapping of chromatin accessibility and DNA methylation within targeted single molecules identifies epigenetic heterogeneity in neural stem cells and glioblastoma[J]. Genome Res,2014,24(2):329-339.
[77]Lopez-Bertoni H,Lal B,Li A,et al.DNMT-dependent suppression of microRNA regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. Oncogene 2014 Oct 20[Epub ahead of print].
[78]Cheng HW,Liang YH,Kuo YL,et al.Identification of thioridazine,an antipsychotic drug,as an antiglioblastoma and anticancer stem cell agent using public gene expression data[J].Cell Death Dis,2015 May 7,6:e1753.doi:10.1038/cddis.2015.77.
[79]Zhou J,Patel TR,Sirianni RW,et al.Highly penetrative,drugloaded nanocarriers improve treatment of glioblastoma[J]. Proc Natl Acad Sci U S A,2013,110(29):11751-11756.
[80]Kast RE,Boockvar JA,Bru¨ning A,et al.A conceptually new treatment approach for relapsed glioblastoma:coordinated undermining of survival paths with nine repurposed drugs(CUSP9)by the International Initiative for Accelerated Improvement of Glioblastoma Care[J]. Oncotarget,2013,4(4):502-530.
[81]Jones PA,Baylin SB.The epigenomics of cancer[J].Cell, 2007,128(4):683-692.
[82]Baylin SB,Esteller M,Rountree MR.Aberrant patterns of DNA methylation,chromatin formation and gene expression in cancer[J].Hum Mol Genet,2001,10(7):687-692.
[83]Christman JK.5-Azacytidine and 5-aza-2-deoxycytidine as inhibitors of DNA methylation:mechanistic studies and their implications for cancer therapy[J].Oncogene, 2002 12,21(35):5483-5495.
[84]So AY,Jung JW,Lee S,et al.DNA meth yltransferase controls stem cell aging by regulating BMI1 and EZH2 through microRNAs[J].PLoS One,May 10 2011,6(5):e19503.
[85]Chang HW,Wang HC,Chen CY.5-azacytidine induces anoikis,inhibits mammosphere formation and reduces metalloproteinase 9 activity in MCF-7 human breast cancer cells[J].Molecules,2014,19(3):3149-3159.
[86]Witt O,Deubzer HE,Milde T,et al.HDAC family:What are the cancer relevant targets?Cancer Lett.,2009,277(1):8-21.
[87]Alexanian AR,Huang YW.Specific combinations of the chromatin-modifying enzyme modulators significantly attenuate glioblastoma cell proliferation and viability while exerting minimal effect on normal adult stem cells growth[J].Tumour Biol 2015 Jun 19.[Epub ahead of print]
[88]Wongtrakoongate P.Epigenetic therapy of cancer stem and progenitor cells by targeting DNA methylation machineries[J].World J Stem Cells,2015,7(1):137-148.
[89]Connolly R,Stearns V.Epigenetics as a therapeutic target in breast cancer[J].J Mammary Gland Biol Neoplasia, 2012,17(3-4):191-204.
[90]Jiang P,Mukthavaram R,Chao Y.Novel anti-glioblastoma agents and therapeutic combinations identified from a collection of FDA approved drugs[J].J Transl Med, 2014,12:13.
[91]Hothi P,Martins TJ,Chen L,et al.High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells[J].Oncotarget,2012,3(10):1124-1136.
[92]Paranjpe A,Zhang R,Ali-Osman F,et al.Disulfiram is a direct and potent inhibitor of human O6-methylguanine-DNA methyltransferase(MGMT)in brain tumor cells and mouse brain and markedly increases the alkylating DNA damage[J].Carcinogenesis,2014,35(3):692-702.
[93]Alvarez AA,Field M,Bushnev S,et al.The effects of histone deacetylase inhibitors on glioblastoma-derived stem cells[J].J Mol Neurosci,2015,55(1):7-20.
[94]Liffers K,Kolbe K,Westphal M,et al.Histone Deacetylase Inhibitors Resensitize EGFR/EGFR v IIIOverexpressing,Erlotinib-Resistant Glioblastoma Cells to Tyrosine Kinase Inhibition[J].Target Oncol 2015 Jun 3.[Epub ahead of print]
[95]Dror N,Mandel M,Lavie G.Unique anti-glioblastoma activities of hypericin are at the crossroad of biochemical and epigenetic events and culminate in tumor cell differentiation[J].PLoS One,2013;16;8(9):e73625.
[96]Xie Q,Mittal S,Berens ME.Targeting adaptive glioblastoma:an overview of proliferation and invasion[J].Neuro Oncol,2014,16(12):1575-1584.
?Prof.Ahmad R.Safa,Indiana University Simon Cancer Center,Indiana University School of Medicine,980 W. Walnut Street,R3-C524,Indianapolis,IN 46202,USA.Tel/Fax:317-278-4952(office)/317-274-8046;E-mail:asafa@iupui.edu.
14 July 2015,Revised 27 July 2015,Accepted 07 August 2015,Epub 20 September 2015
R730.59,Document code:A
The authors reported no conflict of interests.
 THE JOURNAL OF BIOMEDICAL RESEARCH2016年1期
THE JOURNAL OF BIOMEDICAL RESEARCH2016年1期
- THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Molecular docking simulation analysis of the interaction of dietary flavonols with heat shock protein 90
- Myocardin-related transcription factor A cooperates with brahmarelated gene 1 to activateP-selectin transcription
- Assessment of malathion and its effects on leukocytes in human blood samples
- Manifestations of type 2 diabetes in corneal endothelial cell density, corneal thickness and intraocular pressure
- Impact of IL28Bgene polymorphisms rs8099917 and rs12980275 on response to pegylated interferon-α/ribavirin therapy in chronic hepatitis C genotype 4 patients
- Circulating thrombospondin-2 in patients with moderate-to-severe chronic heart failure due to coronary artery disease