
原位合成鈷/還原氧化石墨烯納米粒子催化氨硼烷制氫
2014-09-17 07:00:00楊宇雯盧章輝陳祥樹
物理化學(xué)學(xué)報(bào) 2014年6期
楊宇雯 馮 剛 盧章輝,* 胡 娜 張 飛 陳祥樹,*
(1江西師范大學(xué)化學(xué)化工學(xué)院,南昌330022;2中國石化上海石油化工研究院,上海201208)
1 Introduction
Secure storage and effective release of hydrogen are very important in the application of hydrogen energy.1,2Various hydrogen storage approaches are currently being investigated,including metal hydrides,3sorbent materials,4and chemical hydride systems.5Boron-nitrogen containing compounds have attracted much attention recently for using as hydrogen storage materials due to their suitable thermodynamic and kinetic properties of hydrogen release.6Among them,ammonia borane(NH3BH3,AB)appears to be an appropriate hydrogen storage material because of its high hydrogen content,high stability at room temperature,and nontoxicity.7-9With appropriate catalyst,hydrolysis of AB can release as many as 3 mol of hydrogen per mol of AB.10-12So far a lot of catalysts have been tested for hydrogen generation from the hydrolysis of AB,13-30among which Pt shows the highest activity.17,22,23However,concerning the element abundance and related economic issues,it is a desired goal to prepare low-cost catalysts with high catalytic activity for the terminal practical application of this reaction system in the fuel cell.
Reduced graphene oxide(RGO),a new class two-dimensional carbon nanostructure with one-atom thickness,has many merits such as large theoretical specific surface area,31high intrinsic mobility,32and large density of free electrons,33could be an ideal substrate for growing and anchoring metal NPs.34Up to date,modification of RGO sheets with metallic NPs is mainly synthesized through one-step and two-step methods.35,36The metallic ions and graphene oxide(GO)sheets are reduced at the same time in the former method,while in the latter,GO is firstly reduced and then the metallic ions are deposited on RGO sheets.In the latter way,the complicated reaction steps,long reaction time,and stringent reaction conditions(high temperature,high vacuum,microwave,ultrasound,UV irradiation,etc.)are usually unavoidable.37-39Recently,RGO-supported Ru@Ni,Ag@M(M=Co,Ni,Fe)NPs have been prepared by one-step method under ambient condition and the catalysts exhibit superior catalytic activities.18,21,40However,developing an efficient strategy for one-step in situ synthesis of RGO-supported metal NPs with low-cost and high catalytic activities is still desirable.
Chemical reduction methods provide much greater control over the size and composition,which are widely applied to synthesize metal NPs in solution phase.41This method involves reduction of metal ions in the presence of capping agent using reductant like NaBH4.42When NPs are employed as a catalyst,the capping agent present on the surface diminishes the activity to some extent by blocking some of the active sites.43However,without capping agent,nanoparticles are difficult to synthesize because growth of in situ generated nuclei cannot be halted.Therefore,it is great practical value to synthesis of NPs without using any external capping agent.
Herein,RGO-supported Co NPs were synthesized by using a simple and low-cost one-step approach without using any external capping agent and assistance of high energy.We employed AB itself(much milder than NaBH4)as the reductant during the reactions.The as-synthesized Co/RGO nanocatalysts were used as catalysts in the dehydrogenation and hydrolysis ofAB at room tempertature.
2 Experimental
2.1 Graphite oxide preparation
Graphite oxide was made by a modified Hummers method.44,45Briefly,natural graphite powder(325 mesh)was placed into an 80°C solution of concentrated H2SO4(30 mL),K2S2O8(2.5 g),and P2O5(2.5 g).The mixture was carefully diluted with distilled water,and filtered using a 0.2 micron Nylon Millipore filter to remove the residual acid.The product was dried at 80°C under ambient condition overnight.The pre-oxidized graphite was put into cold concentrated H2SO4,then KMnO4was added gradually under stirring and the temperature of the mixture was kept below 20 °C for 2.5 h.The mixture was stirred at 35 °C for 4 h.Afterwards,250 mL of de-ionized water was added and the suspension was stirred at 100°C for another 2 h.Subsequently,additional 300 mL of de-ionized water was added.Shortly after that,7 mL of 30%(w)H2O2was added to the mixture to terminate the reaction.The suspension was then repeatedly centrifuged and washed first with 5%(w)HCl solution and then with water.Exfoliation of graphite oxide to GO was achieved by ultrasonication of the dispersion for 30 min.46
2.2 In situ synthesis of Co/RGO catalysts and their catalytic studies of hydrolytic dehydrogenation of AB
8 mL aqueous solution containing CoCl2(24.03 mg)and GO solution(1.07 g,containing 0.412%(w)GO)was kept in a 25 mL two-necked round-bottom flask.One neck was connected to a gas burette,and the other was connected to a pressureequalization funnel to introduce 2 mL of aqueous solution con-taining 34.3 mg(1 mmol)AB.The reactions were started when the aqueous AB solution was added to the flask with vigorously stirring.The evolution of gas was monitored using the gas burette.After the hydrogen generation reaction was completed,34.3 mg(1 mmol)AB was added to the flask,the evolution of gas was monitored.A water bath was used to control the temperature of the reaction solution(the amount of AB in the processes of in situ synthesis of Co/RGO catalyst and hydrolytic dehydrogenation are the same,1 mmol AB was used as reductant in the first process and another 1 mmol AB was used for the hydrolytic dehydrogenation test).
For comparision,GO and Co NPs were synthesized using AB as reductant,RGO and Co/RGO were synthesized using NaBH4as reductant.The as-synthesized catalysts were used for the hydrolysis ofAB.
2.3 Kinetic studies of hydrolytic dehydrogenation of AB catalyzed by Co/RGO
In order to establish the rate law for catalytic hydrolysis of AB using Co/RGO as catalyst,three different sets of experiments were performed in the same way described in Section 2.2.In the first set of experiment,the different concentrations of Co(0.04,0.06,0.08,and 0.10 mmol)were performed at room temperature(25°C)while the AB concentration was kept the same(1 mmol).In the second set of experiment,the different concentrations of AB(1.0,1.5,2.0,and 2.5 mmol)were performed at room temperature(25°C)while the Co concentration was kept the same(0.1 mmol).Finally,temperature was varied at 25,30,35,and 40°C while the molar ratio of metal/AB(0.1 mmol Co and 1 mmol AB)was kept constant of 0.1 to obtain the activation energy(Ea).
2.4 Stability test
For stability test,catalytic reactions were repeated 5 times by adding other equivalent of AB(1 mmol)into the mixture after the previous cycle.The molar ratio of metal/AB was kept at 0.1.
2.5 Catalyst characterization
Transmission electron microscope(TEM),energy-diepersive X-ray spectroscopy(EDS),and selected area electron diffraction(SAED)were observed using FEI Tecnai G20 U-Twin TEM instrument operating at 200 kV.Powder X-ray diffraction(XRD)studies were performed on a Rigaku RINT-22005 X-ray diffractometer with a Cu Kαsource(40 kV,20 mA).X-ray photoelectron spectroscopy(XPS)measurement was performed with a Thermo ESCALAB 250XI multifunctional imaging electron spectrometer.Fourier transform infrared(FTIR)spectra were collected at room temperature by using a Thermo Nicolet 870 instrument using KBr discs in the 500-4000 cm-1region.Raman spectrometer was carried out using a confocal Raman microscope(LabRAM HR).
3 Results and discussion
3.1 Synthesis and characterization
As well known,the Co(II)cations were difficult to reduce to Co by AB(a mild reducing agent)at room temperature,10,14which is also evidenced in the present experiments(Fig.S1(see Supporting Information)).Interestingly,in the presence of GO,the Co(II)cations could be reduced to Co by using AB as a reductant within a short period(Fig.S1).The decrease of induction period may result from the charge transfer across the graphene oxide-cobalt interface due to the graphene oxide-cobalt spacing and Fermi lever difference.21The RGO-supported Co(Co/RGO)NPs were successfully synthesized by reducing a mixture containing CoCl2and GO with AB as the sole reductant.The microstructures of the samples were characterized by TEM,high-resolution TEM(HRTEM),EDS,and SAED(Fig.1).As shown in Fig.1(a),the GO sheets are transparent and corrugated together.The TEM images of Co/RGO(Fig.1(b,c))show that most of the Co NPs lay flat on the RGO.Moreover,the aggregation of Co NPs was found in Co/RGO,which could be due to the magnetic property of Co NPs.The EDS spectrum of the specimen shows the presence of Co(Fig.S2,which was taken from the specially marked area in the TEM image(Fig.1(c)).A close examination of the catalysts by HRTEM(Fig.1(d)),the d-spacing of the particle lattice is~0.204 nm,which is consistent with the SAED pattern(4.9 nm-1in Fig.1(d)inset)and the(111)plane of cubic Co(JCPDS No.15-0806).Moreover,the corresponding SAED pattern demonstrates the low degree of crystallinity of Co.
Fig.2 shows the powder XRD patterns of GO and Co/RGO.The diffraction peak at around 44.23°attributed to Co(111)is observed in Co/RGO,which is consistent with the HRTEM result(Fig.1(d)).Furthermore,the most intense peak at around 11.5°corresponding to the(001)reflection of GO disappeared,while a new peak at around 24.58°was observed in Co/RGO,indicating that GO is successfully reduced to the RGO.
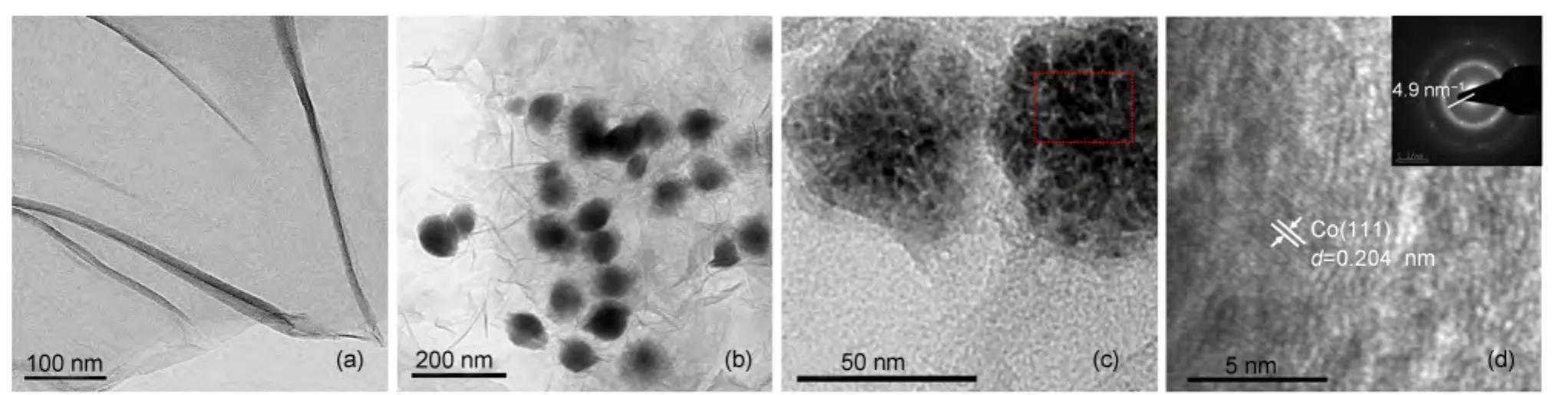
Fig.1 (a)TEM images of GO;(b,c)TEM images of Co/RGO nanocatalysts;(d)HRTEM image of Co/RGO nanocatalysts and SAED pattern(inset)
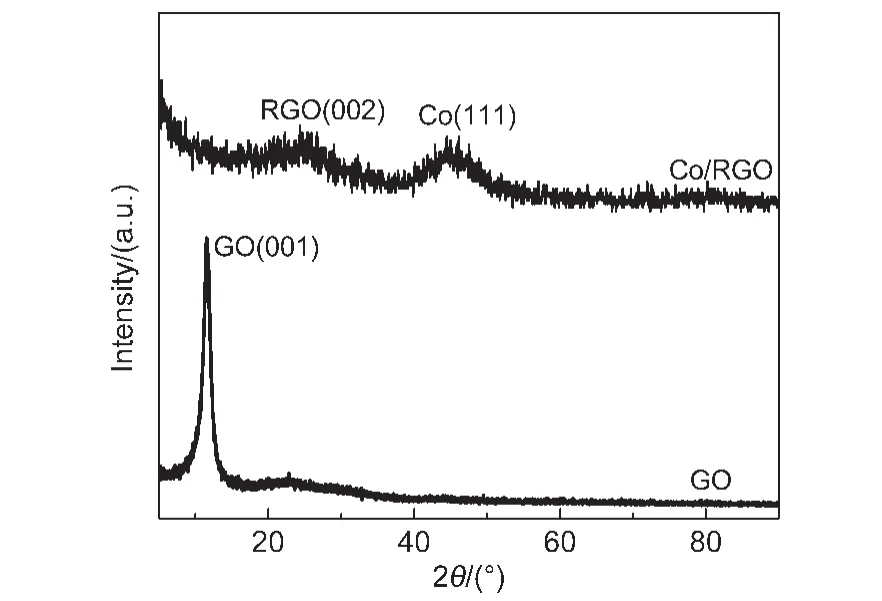
Fig.2 XRD patterns of GO and Co/RGO nanocatalyst
Co/RGO was further characterized by XPS to investigate the surface nature of the Co NPs and RGO(Fig.3).Compared with the peaks of GO(Fig.3(A)),the intensities of oxygen containing functional groups(such as―C―O,―C=O,―COO)in Co/RGO(Fig.3(B))decrease significantly,also revealing the reduction of GO to RGO.Fig.3(C)shows the peaks of Co 2p.The peak at 778.5 eV stands for Co0,the two peaks at 780.6 and 786.7 eV stand for oxidized Co.The formation of the oxidized Co most likely occurs during the sample preparation process for XPS measurements.The presence of carbon-oxygen bonding and oxidized Co are also evident in the O 1s spectrum of Co/RGO(Fig.3(D)).The O 1s spectrum shows peaks at 534.0,533.0,531.6,and 530.4 eV,which could be assigned to―COO,―C―O,―C=O,oxidized Co.

Fig.3 XPS spectra of C 1s of(A)GO and(B)Co/RGO,(C)Co 2p of Co/RGO,and(D)O 1s of Co/RGO
As shown in Fig.4(a),two peaks centered at 1316.92 and 1584.57 cm-1appear in the Raman spectra of the GO and Co/RGO,corresponding to the D and G bands of the carbon products,respectively.The D band is an indication of disorder of GO originating from defects associated with vacancies,grain boundaries,and amorphous carbon species,while the G band is ascribed to the E2gphonon of C sp2atoms in a 2-dimensional hexagonal lattice.The peak intensity ratio of the D to G band(ID/IG)is generally accepted to reflect the degree of graphitization of carbonaceous materials and defect density.After loading of Co NPs,the ID/IGof GO is increased from 1.2 to 1.6.The relative changes in the D to G peak intensity ratio also confirm the reduction of GO during the in situ fabrication.
Fig.4(b)shows the FTIR spectra of GO and Co/RGO.As for the FTIR spectrum of GO,the broad and intense band at 3401.9 cm-1is ascribed to the stretching of O―H.The weak band at 1723.6 cm-1is assigned to C=O stretching vibration in carbonyl or carboxylic groups.The peak at 1621.6 cm-1is pertinent to the vibrations of the absorbed water molecules and the skeletal vibration of unoxidized graphitic domains.The bands at 1399.2 and 1074.4 cm-1are associated with the O―H vibration in carboxyl acid and the deformation of the C―O band,respectively.After the formation of Co/RGO,the disappearance of C=O at 1723.6 cm-1,C―OH peak at 1399.2 cm-1,and the C―O peak at 1074.4 cm-1of GO further indicates that GO was reduced to RGO during the process.
3.2 Catalytic activities for hydrolysis of AB
As shown in Fig.5,no hydrogen generation was observed for GO and RGO,suggesting that GO and RGO have no catalytic activity for the hydrolysis of AB.The as-synthesized Co/RGO generates a stoichiometric amount of hydrogen(H2/NH3BH3molar ratio:3.0)in 4.37 min with a turnover frequency(TOF)value of 6.86 mol·mol-1·min-1.The as-synthesized Co/RGO nanocatalysts display much better catalytic activities than pure Co NPs.The enhanced catalytic activity of Co/RGO for AB hydrolysis reaction should result from the cooperative effect between RGO and Co NPs,which is mainly caused by the strongly interfacial interaction between RGO and Co NPs during the catalytic process.27Compared with pre-catalysts reduced by NaBH4(Co/RGO(SB)),the as-synthesized nanocatalysts generated by AB(Co/RGO(AB))exhibit a superior catalytic activity(Fig.5 and Fig.S3),indicating that AB can be used as both a potential hydrogen storage material and an efficient reducing agent.
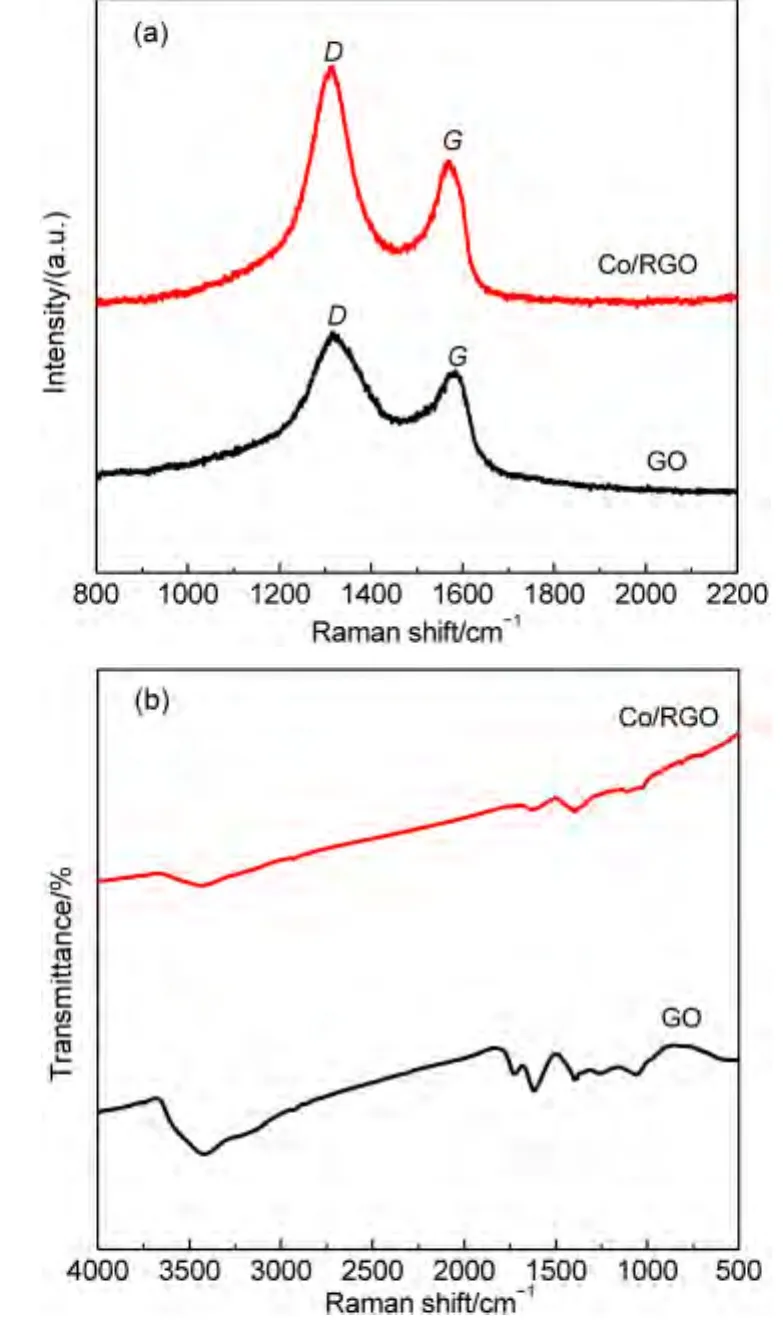
Fig.4 (a)Raman and(b)FTIR spectra of the GO and Co/RGO
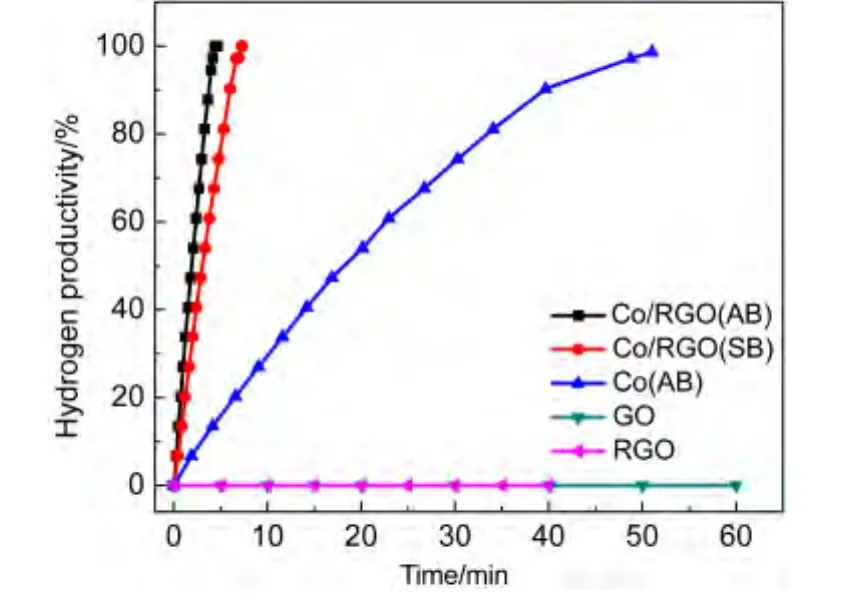
Fig.5 Plots of hydrogen productivity vs time for hydrolysis of ammonia borane(0.10 mol·L-1,10 mL)catalyzed by Co/RGO reduced byAB and NaBH4(SB)respectively,Co NPs reduced byAB,GO,and RGO
Fig.S4(a)shows the plots of hydrogen generation from the hydrolysis of AB solution in the presence of different Co/RGO concentration at 25°C.The initial rate of hydrogen generation was determined from the initial nearly linear portion of each plot.Fig.S4(b)shows the plot of hydrogen generation rate versus Co/RGO concentration in a logarithmic scale.A slope of 1.05 in the inset indicates that the hydrolysis reaction catalyzed by Co/RGO is first-order in catalyst concentration.
The effect of substrate concentration on the hydrogen generation rate was also studied by performing a series of experiments starting with different initial concentrations of AB while keeping the catalyst concentration at 10 mmol·L-1Co at room temperature(Fig.6).It can be clearly concluded by the slope of the line in Fig.6(b)that the hydrogen generation rate from the catalytic hydrolysis of AB is practically independent from AB concentration.In other words,the hydrolysis of AB catalyzed by Co/RGO is zero order with respect to the substrate concentration.

Fig.6 Plots of(a)volume of hydrogen generated vs time,(b)hydrogen generation rate versus the concentration ofAB(both in logarithmic scale)

Fig.7 (a)Plots of volume of hydrogen generated vs time for Co/RGO catalyzed hydrolysis ofAB at different temperatures(nCo/nAB=0.1);(b)Arrhenius plot obtained from the data of Fig.7(a)
In order to get the activation energy(Ea)of the hydrolysis of AB catalyzed by Co/RGO,the hydrolytic reactions at the temperature range of 298-313 K were carried out.The values of rate constant k at different temperatures were calculated from the slope of the linear part of each plot from Fig.7(a).The Arrhenius plot of lnk vs 1/T for the catalyst is plotted in Fig.7(b),from which the apparent activation energy was determined to be approximately 27.10 kJ·mol-1,being lower than most ofthe reported Eavalues(Table 1),indicating the superior catalytic performance of the as-synthesized Co/RGO nanocatalysts.
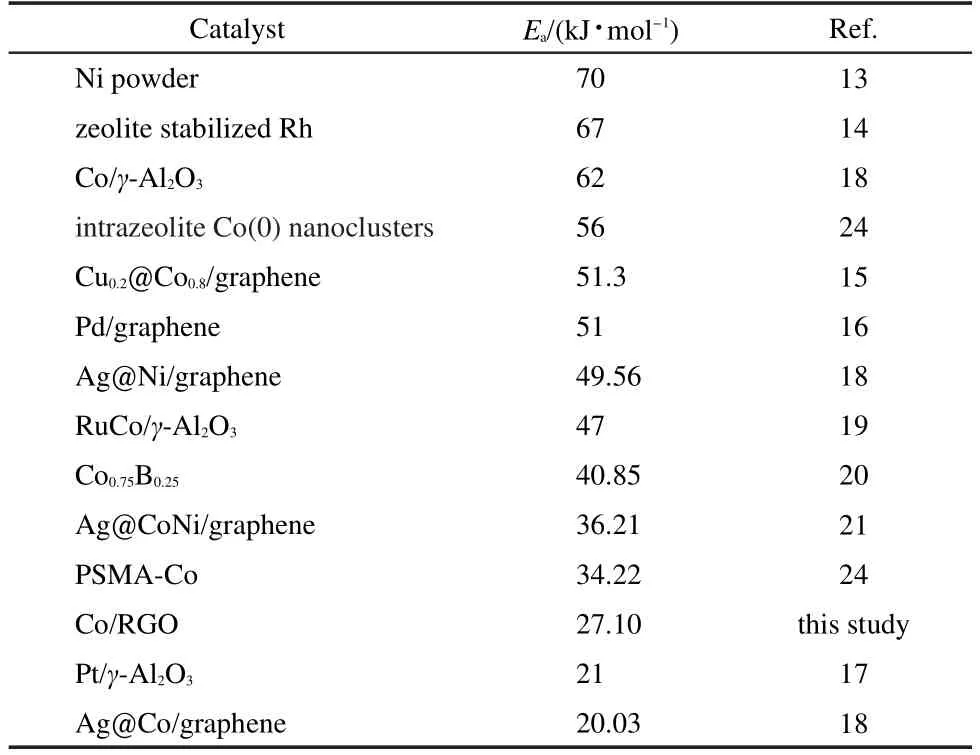
Table 1 Values of activation energy(Ea)for hydrolysis ofAB catalyzed by different catalysts
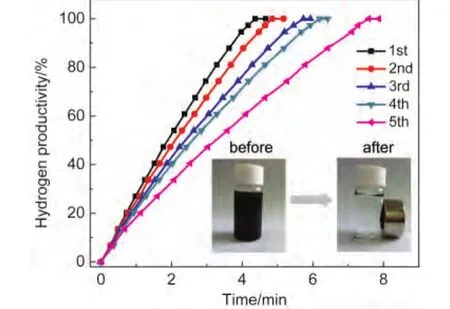
Fig.8 Plots of hydrogen productivity vs time for Co/RGO catalyzed hydrolysis ofAB(0.10 mol·L-1,10 mL)from 1st to 5th cycles(nCo/nAB=0.1)
3.3 Reusability and recycle ability
The reusability is of great importance for the practical application of catalyst.The recyclability of Co/RGO nanocatalyst up to the fifth run for hydrolysis of AB is shown in Fig.8.The complete release of hydrogen is achieved in each of the subsequent catalytic runs in the hydrolysis of AB catalyzed by Co/RGO nanocatalysts.This indicates that Co/RGO can be repeatedly used as active catalyst in the hydrolysis of AB.The observed decrease in catalytic activity in subsequent runs may be attributed to the passivation of nanocatalyst surface by the precipitation of metaborate products.22Moreover,the in situ synthesized Co/RGO nanocatalysts are magnetic and thus can be separated from the reaction solution by an external magnet(inset in Fig.8),which makes the practical recycling application of nanocatalysts more convenient.
4 Conclusions
In summary,we have developed a facial in situ one-step method for the synthesis of magnetic RGO-supported Co NPs using AB as the sole reductant.The as-synthesized nanocatalysts exhibit a high catalytic activity for hydrolytic dehydrogenation of AB with the activation energy Eaof 27.10 kJ·mol-1,which is lower than most of the reported data for the same reaction using non-noble metal catalysts and even some noble metal containing catalysts.Moreover,the Co/RGO nanocatalysts show good durable stability and magnetically recyclability for the hydrolytic dehydrogenation of AB due to the magnetic property from Co,which makes the practical recycling application of the catalyst more convenient.This simple synthetic method can be extended to the other RGO-based metallic systems for more application.
Supporting Information: available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1) Schlapbach,L.;Züttel,A.Nature 2001,414,353.doi:10.1038/35104634
(2)Grochala,W.;Edwards,P.P.Chem.Rev.2004,104,1283.doi:10.1021/cr030691s
(3) Graetz,J.Chem.Soc.Rev.2009,38,73.doi:10.1039/b718842k
(4)Suh,M.P.;Park,H.J.;Prasad,T.K.;Lim,D.W.Chem.Rev.2012,112,782.doi:10.1021/cr200274s
(5) Staubitz,A.;Robertson,A.P.M.;Manners,I.Chem.Rev.2010,110,4079.doi:10.1021/cr100088b
(6) Chen,P.;Zhu,M.Mater.Today 2008,11,36.
(7) Lu,Z.H.;Xu,Q.Funct.Mater.Lett.2012,5,1230001.doi:10.1142/S1793604712300010
(8)Yadav,M.;Xu,Q.Energy Environ.Sci.2012,5,9698.doi:10.1039/c2ee22937d
(9)Lu,Z.H.;Yao,Q.L.;Zhang,Z.J.;Yang,Y.W.;Chen,X.S.J.Nanomater.2014,729029.
(10) Rakap,M.;Kalu,E.E.;?zkar,S.J.Power Sources 2012,210,184.doi:10.1016/j.jpowsour.2012.03.025
(11)Yan,J.M.;Wang,Z.L.;Wang,H.L.;Jiang,Q.J.Mater.Chem.2012,22,10990.doi:10.1039/c2jm31042b
(12)Yang,Y.W.;Zhang,F.;Wang,H.L.;Yao,Q.L.;Chen,X.S.;Lu,Z.H.J.Nanomater.2014,294530.
(13)Cheng,F.Y.;Ma,H.;Li,Y.M.;Chen,J.Inorg.Chem.2007,46,788.doi:10.1021/ic061712e
(14) Basu,S.;Brockman,A.;Gagare,P.;Zheng,Y.;Ramachandran,P.V.;Delgass,W.N.;Gore,J.P.J.Power Sources 2009,188,238.doi:10.1016/j.jpowsour.2008.11.085
(15)Du,Y.S.;Cao,N.;Yang,L.;Luo,W.;Cheng,G.Z.New J.Chem.2013,37,3035.doi:10.1039/c3nj00552f
(16)Xi,P.X.;Chen,F.J.;Xie,G.Q.;Ma,C.;Liu,H.Y.;Shao,C.W.;Wang,J.;Xu,Z.H.;Xu,X.M.;Zeng,Z.Z.Nanoscale 2012,4,5597.doi:10.1039/c2nr31010d
(17) Chandra,M.;Xu,Q.J.Power Sources 2007,168,135.doi:10.1016/j.jpowsour.2007.03.015
(18)Yang,L.;Luo,W.;Cheng,G.E.ACS Appl.Mater.Interfaces 2013,5,8231.doi:10.1021/am402373p
(19) Rachiero,G.P.;Demirci,U.B.;Miele,P.Int.J.Hydrog.Energy 2011,36,7051.doi:10.1016/j.ijhydene.2011.03.009
(20)Simagia,V.I.;Komova,O.V.;Ozerova,A.M.;Netskina,O.V.;Odegova,G.V.;Kelleman,D.G.;Bulavcheoko,O.V.;Ishchenko,A.V.Appl.Catal.A:Gen.2011,384,86.
(21)Yan,L.;Su,J.;Meng,X.Y.;Luo,W.;Cheng,G.Z.J.Mater.Chem.A 2013,1,10016.doi:10.1039/c3ta11835e
(22) Lu,Z.H.;Li,J.P.;Zhu,A.L.;Yao,Q.L.;Huang,W.;Zhou,R.Y.;Zhou,R.F.;Chen,X.S.Int.J.Hydrog.Energy 2013,38,5330.doi:10.1016/j.ijhydene.2013.02.076
(23)Lu,Z.H.;Jiang,H.L.;Yadav,M.;Aranishi,K.;Xu,Q.J.Mater.Chem.2012,22,5065.doi:10.1039/c2jm14787d
(24) Rakap,M.;?zkar,S.Int.J.Hydrog.Energy 2010,35,3341.doi:10.1016/j.ijhydene.2010.01.138
(25) Metin,?.;?zkar,S.Int.J.Hydrog.Energy 2011,36,1424.
(26)Yao,Q.L.;Shi,W.M.;Feng,G.;Lu,Z.H.;Zhang,X.L.;Tao,D.J.;Kong,D.J.;Chen,X.S.J.Power Sources 2014,257,293.doi:10.1016/j.jpowsour.2014.01.122
(27)Yang,Y.W.;Lu,Z.H.;Hu,Y.J.;Zhang,Z.J.;Shi,W.M.;Chen,X.S.;Wang,T.T.RSC Advances 2014,4,13749.doi:10.1039/c3ra47023g
(28) Chandra,M.;Xu,Q.J.Power Sources 2006,156,190.doi:10.1016/j.jpowsour.2005.05.043
(29) Rakap,M.;Kalu,E.E.;?zkar,S.Int.J.Hydrog.Energy 2011,36,1448.doi:10.1016/j.ijhydene.2010.10.097
(30)Eom,K.S.;Cho,K.W.;Kwon,H.S.Int.J.Hydrog.Energy 2010,35,181.
(31) Garaj,S.;Hubbard,W.;Reina,A.;Kong,J.;Branton,D.;Golovchenko,J.A.Nature 2010,467,190.doi:10.1038/nature09379
(32) Lee,C.;Wei,X.D.;Kysar,J.W.;Hone,J.Science 2008,321,385.doi:10.1126/science.1157996
(33)Choi,B.G.;Hong,J.;Park,Y.C.;Jung,D.H.;Hong,W.H.;Hammond,P.T.;Park,H.S.ACS Nano 2011,5,5167.doi:10.1021/nn2013113
(34) Hu,Y.J.;Jin,J.;Zhang,H.;Wu,P.;Cai,C.X.Acta Phys.-Chim.Sin.2010,26(8),2073.[胡耀娟,金 娟,張 卉,吳 萍,蔡稱心.物理化學(xué)學(xué)報(bào),2010,26(8),2073.]doi:10.3866/PKU.WHXB20100812
(35)Li,S.M.;Wang,B.;Liu,J.H.;Yu,M.;An,J.W.Acta Phys.-Chim.Sin.2012,28(11),2754.[李松梅,王 博,劉建華,于 美,安軍偉.物理化學(xué)學(xué)報(bào),2012,28(11),2754.]doi:10.3866/PKU.WHXB201208292
(36)Li,Y.X.;Wei,Z.D.;Zhao,Q.L.;Ding,W.;Zhang,Q.;Chen,S.G.Acta Phys.-Chim.Sin.2011,27(4),858.[李云霞,魏子棟,趙巧玲,丁 煒,張 騫,陳四國.物理化學(xué)學(xué)報(bào),2011,27(4),858.]doi:10.3866/PKU.WHXB20110411
(37)Mazumder,V.;Chi,M.F.;More,K.L.;Sun,S.H.Angew Chem.Int.Edit.2010,49,9368.doi:10.1002/anie.201003903
(38) Vinodgopal,K.;Neppolian,B.;Lightcap,I.V.;Grieser,F.;Ashokkumar,M.;Kamat,P.V.J.Am.Chem.Soc.2010,1,1987.
(39)Liu,C.B.;Wang,K.;Luo,S.L.;Tang,Y.H.;Chen,L.Y.Small 2011,7,1203.doi:10.1002/smll.v7.9
(40)Cao,N.;Su,J.;Luo,W.;Cheng,G.Z.Int.J.Hydrog.Energy 2014,39,426.doi:10.1016/j.ijhydene.2013.10.059
(41) Roucoux,A.;Schulz,J.;Patin,H.Chem.Rev.2002,102,3757.doi:10.1021/cr010350j
(42)Yang,L.;Cao,N.;Du,C.;Dai,H.M.;Hu,K.;Luo,W.;Cheng,G.Z.Materials Letters 2014,115,113.doi:10.1016/j.matlet.2013.10.039
(43)Astruc,D.;Lu,F.;Aranzaes,J.R.Angew Chem.Int.Edit.2005,44,7852.
(44)Hummers,W.S.;Offeman,R.E.J.Am.Chem.Soc.1958,80,1339.doi:10.1021/ja01539a017
(45) Kovtyukhova,N.I.;Ollivier,P.J.;Martin,B.R.;Mallouk,T.E.;Chizhik,S.A.;Buzaneva,E.V.;Gorchinskiy,A.D.Chem.Mater.1999,11,771.doi:10.1021/cm981085u
(46) Chen,H.Q.;Müller,M.B.;Gilmore,K.J.;Wallace,G.G.;Li,D.Adv.Mater.2008,20,3557.doi:10.1002/adma.200800757
猜你喜歡
當(dāng)代音樂(2024年5期)2024-06-03 15:19:37
無機(jī)化學(xué)學(xué)報(bào)(2024年1期)2024-01-20 03:55:50
化工職業(yè)技術(shù)教育(2021年5期)2021-11-09 03:10:40
化工職業(yè)技術(shù)教育(2021年1期)2021-03-15 06:57:44
作文周刊·小學(xué)一年級版(2017年11期)2017-06-20 00:16:48
作文周刊·小學(xué)一年級版(2016年48期)2017-06-06 22:16:37
作文周刊·小學(xué)一年級版(2016年40期)2017-03-03 12:41:57
High Technology Letters(2016年2期)2016-12-05 01:31:30
化工學(xué)報(bào)(2016年3期)2016-03-14 08:37:00
瘋狂英語(雙語世界)(2016年3期)2016-02-27 10:11:43
