
Kinetics Study on O2Adsorption and OHadDesorption at Pt(111),Its Implication to Oxygen Reduction Reaction Kinetics
2014-07-19 11:18:24FanYangLingwenLiaoMingfangLiDongMeiYanxiaChen
Fan Yang,Ling-wen Liao,Ming-fang Li,Dong Mei,Yan-xia Chen
Hefei National Laboratory for Physical Sciences at the Microscale,Department of Chemical Physics, University of Science and Technology of China,Hefei 230026,China
Kinetics Study on O2Adsorption and OHadDesorption at Pt(111),Its Implication to Oxygen Reduction Reaction Kinetics
Fan Yang,Ling-wen Liao,Ming-fang Li,Dong Mei,Yan-xia Chen?
Hefei National Laboratory for Physical Sciences at the Microscale,Department of Chemical Physics, University of Science and Technology of China,Hefei 230026,China
Kinetics of dissociative O2adsorption,OHaddesorption,and oxygen reduction reaction (ORR)at Pt(111)electrode in 0.1 mol/L HClO4has been investigated.Reversible OHadadsorption/desorption occurs at potentials from 0.6 V to 1.0 V(vs.RHE)with the exchange current density of ca.50 mA/cm2at 0.8 V,the fast kinetics of OHaddesorption indicates that it should not be the rate determining step for ORR.In the kinetic-or kinetic-mass transport mix controlled potential region,ORR current at constant potential displays slight decrease with reaction time.ORR current in the positive-going potential scan is slightly larger than that in the subsequent negative-going scan with electrode rotation speed(>800 r/min)and slow potential scan rate(<100 mV/s).The open circuit potential of Pt/0.1 mol/L HClO4interface increases promptly from 0.9 V to 1.0 V after switch from O2free-to O2-saturated solution.The increase of open circuit potential as well as ORR current decays under potential control due to the accumulation of OHadfrom dissociative adsorption of O2.It indicates that at Pt(111)the net rate for O2decomposition to OHadis slightly faster than that for OHadremoval,one cannot simply use the assumption of rate determining step to discuss ORR kinetics.Instead,the ORR kinetics is determined by both the kinetics for O2decomposition to OHadas well as the thermo-equilibrium of OHad+H++e?H2O.
Oxygen reduction reaction,Pt(111)electrode,Rate determining step,Kinetics, Overpotential,Thermodynamic equilibrium
I.INTRODUCTION
Oxygen reduction reaction(ORR)is one of the most important model systems in electrocatalysis.Uncovering factors which limit ORR kinetics is paramount to design better electrocatalysts for fuel cells.Pt and Pt-based materials are the most active ORR electrocatalysts,but even on the best Pt based electrocatalysts large overpotential(η>0.22 V)are still required in order to achieve any appreciable ORR current[1-10].In acidic solutions,the overall reaction for ORR is:
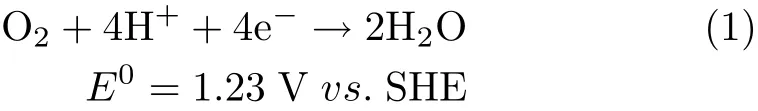
On Pt-based electrodes,two pathways are suggested for ORR:one major path with 4e reduction of O2via a direct or series pathway,and a minor route involving the‘2e’pathway to peroxide when there is strongly adsorbed species on Pt electrode[11-13].On the Pt based electrocatalysts,intermediates such as O22-, O2-,HO2-,H2O2,O,and OH may be produced[10]. Most of these intermediates are hard to be detected by experimental methods due to their short lifetime and their low coverage at the electrode surface.As a result, it will be very difficult to derive the detailed mechanism for ORR.Even,by now there is no consensus on its rate determining step(r.d.s.)at all.Reactions(2)-(5) listed below have all been suggested to be the r.d.s.for ORR in acidic electrolyte[2,3,5,6]:
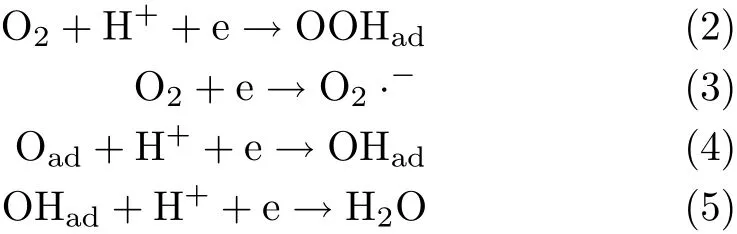
In order to clarify the above discrepancies and to discover the key factors which limit ORR kinetics at Pt, we have studied the kinetics for O2adsorption,OHaddesorption and ORR at Pt(111)electrode in 0.1 mol/L HClO4.Kinetic implication for ORR at Pt-based electrocatalysts will be brief l y discussed based on present results and density functional theory(DFT)calculations.
II.EXPERIMENTS
Experimental conditions are similar to that described in Ref.[7].0.1 or 1.0 mol/L HClO4solution was pre-pared using 70%perchloric acid(Suprapure,Sigma-Aldrich)and ultra-pure water(18.2 M?/cm),N2and O2were of purity 99.999%(Linde China).Most measurements were done under hanging meniscus rotating disk electrode conf i guration in a conventional three electrodes cell.Bead type Pt(111)was used as a working electrode.A reversible hydrogen electrode(RHE) and a Pt wire were used as reference and counter electrode.All potentials were quoted against the RHE. Reaction kinetics for O2,OHadadsorption/desorption and ORR were examined using potential step technique at 2000 r/min and fast scan cyclic voltammetry at 3600 r/min after purging the solution with O2for 20 min,the high electrode rotation speed was used in order to eliminate the interference of mass transport effect.The change of OCP for Pt when switching from O2free to O2saturated 0.1 mol/L HClO4solution was carried out with a f l ow cell[14].
III.RESULTS AND DISCUSSION
CVs for Pt(111)electrode in 0.1 mol/L HClO4solution are given in Fig.1.From Fig.1(a),it is noticed that there is symmetric butter fl y feature in the potential region from 0.4 V to 0.85 V with a sharp peak at 0.8 V.It is followed by a current plateau in the potential region from 0.85 V to 1.0 V,which is obviously higher than that in the double layer potential region.The“butterfl y”feature in the potential region of 0.5 V<E<0.85 V together with the small plateau from 0.85 V to 1.0 V is associated with the adsorption of OHadfrom water and its desorption[15].
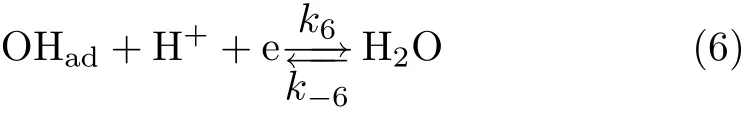
Irreversible Pt-Oxformation/reduction only occurs when the upper potential limit used for the cyclic voltammetry is higher than 1.0 V(Fig.1(b)).A typical polarization curve for ORR recorded in O2saturated 0.1 mol/L HClO4solution at 2000 r/min is given in Fig.1(d).By comparing the base CV recorded in O2-free solution,it is noticed that ORR only occurs at potentials negative of the upper limit(ca.0.98 V) where reversible OHadadsorption/desorption occurs [7].The kinetic and kinetic-mass transport mixcontrolled potential regions for ORR at Pt(111)just locate in the same potential region where OHadadsorption/desorption is reversible in O2-free solution.Systematic studies on ORR at Pt(111)in solutions with other pH reveal that such phenomenon applies for all solutions with pH within the range of 1≤pH≤13 and ORR kinetics at Pt(111)ispH independent[7].Theequilibrium potential for Eq.(6)is:
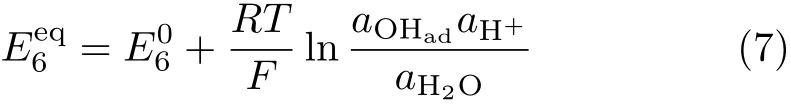
where a is the activity of respective species specified in the subscript,E06is standard potential of reaction(6), R is molar gas constant,T is temperature,F is Faraday constant.The superimposition of the onset potential for ORR with that of OHaddesorption indicates that the high value of the overpotential at the onset for ORR at Pt(111)is determined by.This can also be understood by the fact that since reaction(5)(i.e.,the forward reaction in reaction(6))is the last step for ORR, hence ORR only occurs when E<.This conclusion agrees well with recent predictions based on DFT calculation,i.e.,among all possible elementary steps for ORR at Pt(111),OHaddesorption has the most positive Gibbs free energy change(?G),which renders the onset potential for ORR ca.0.25 V negative of 1.23 V [6].Reaction(5)is called the potential determining step (p.d.s.)for ORR at Pt(111)[6,9].
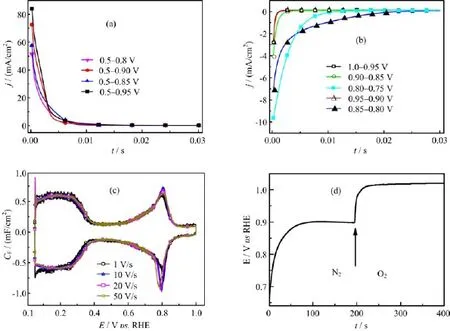
FIG.2 Current transients for(a)OHadformation and(b)OHaddesorption at Pt(111)in 0.1 mol/L HClO4after potential step to the respective potentials.(c)Capacitance CFvs.E curves for Pt(111)in 0.1 mol/L HClO4at various potential scan rates.(d)The change of open circuit potential for Pt/0.1 mol/L HClO4interface after switching from a O2-free to O2-saturated solution.
To clarify whether OHaddesorption or the dissociative adsorption of O2is the r.d.s for ORR at Pt(111), fast scan voltammetry and potential step experiments were carried out to derive the kinetics of the respective processes.Figure 2 displays the current transients for OHadformation and the reductive removal of OHadat Pt(111)in 0.1 mol/L HClO4after stepping from 0.5 V to the respective reaction potentials or stepping from higher potential to lower ones.From Fig.2 it is seen that in O2free solution OHadestablishes its equilibrium coverage within ca.5 ms to 30 ms after potential step from either higher or lower potentials to the desired ones.This reveals that the kinetics for both OHadadsorption and desorption are very fast,which is in well agreement with literature report[16-18].The jt curves recorded during the potential step experiment can be described as[19]:
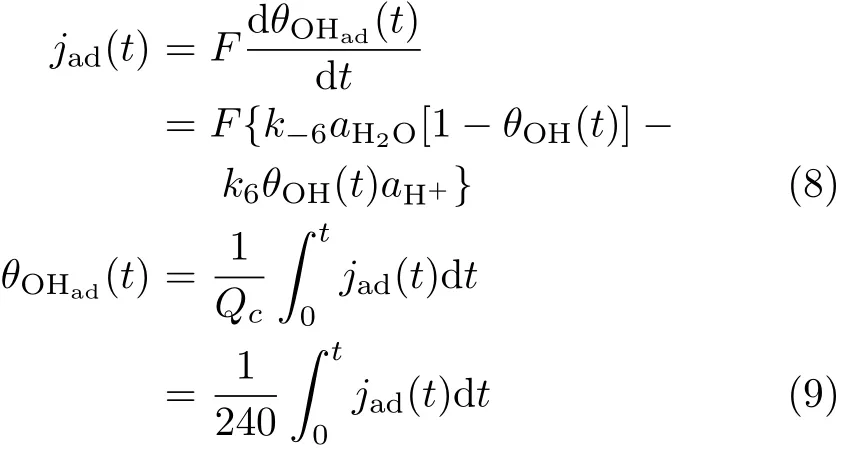
where θOHadis coverage of OH species.From the current transients and Eq.(8)and Eq.(9)the rate constant for OHadadsorption/desorption(k6,k-6)can be derived(note only data recorded after ca.0.3 ms have been taken into estimation,cell time constant is τ=90μs.By def i ning the state with θOHad=0.3 ML at 0.8 V as the standard state,exchange current density j0for reaction(6)is estimated as follows:
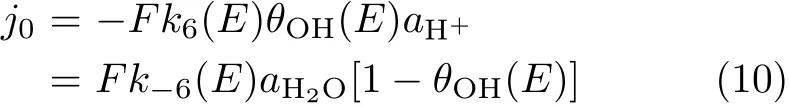
At 0.8 V,j0is estimated to be ca.50 mA/cm2.The current density for OHaddesorption(the forward reaction in reaction(6))are estimated to be-2.7 and -7.1 mA/cm2at 0.95 and 0.9 V by assuming a symmetric factor of 0.5.The fast kinetics for OHadadsorption/desorption is further supported by the good symmetry for OHadadsorption/desorption under fast potential scan rate(Fig.2(c)).On the other hand,the kinetic current densities(jk,ORR)for ORR after elimination of the mass transport effect are-0.35 and-1.8 mA/cm2at 0.95 and 0.90 V,respectively,as derived from the Koutecky-Levich equation[8].Obviously, jk,ORRis smaller than the rate for OHaddesorption under otherwise identical condition,indicating that the kinetics for ORR is not limited by the“slow”kinetics for OHaddesorption.In addition,reaction(4)should also not be the r.d.s.for ORR based on the Br?nsted-Evans-Polanyi relationship and it has similar Gibbs free energy change as that for reaction(5)[6].
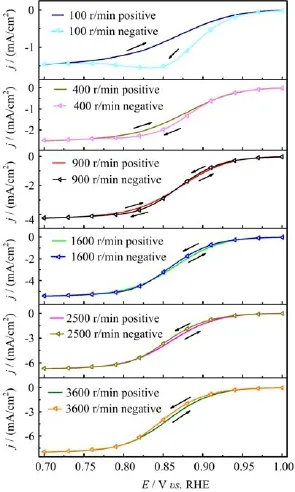
FIG.3 Positive-and negative-going scan polarization curves for ORR at Pt(111)in O2saturated 0.1 mol/L HClO4with different electrode rotation speed,potential scan rate of 50 mV/s.
On the other hand,the Tafel slope for E vs.lgjkis ca.-70 mV/dec in the potential region from 0.95 V to 0.85 V(Fig.1(d))[7].When normalized to the active surface sites by taking into account that OHadcoverage is the same as that in O2free solution(which can be determined from the OHadadsorption charge in the potential region from 0.6 V to 1.0 V based on the CV given in Fig.1(a)),the Tafel slope is just ca.-78 mV/dec (Fig.1(e)).This indicates that the OH coverage does not affect the Tafel slope significantly.Assuming that OHad|Oadcoverage under ORR condition is higher than that in O2-free solution under otherwise identical condition and the discrepancies for OHadcoverage in these two cases increase with increase in electrode potential, the Tafel slope after being normalized with the free active sites should be within the range of-70 mV/dec to -78 mV/dec.This value is significantly smaller than -120 mV/dec,which indicates that the f i rst charge transfer step(either reactions(2)or(3))is probably not the r.d.s for ORR on Pt(111)at E>0.85 V.
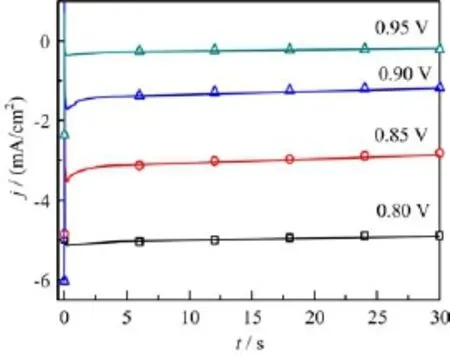
FIG.4 Current transients for ORR at Pt(111)in 0.1 mol/L HClO4after stepping from 0.5 V to the respective reaction potentials as indicated,electrode rotation speed is 2000 r/min.
Another evidence which supports fast decomposition of O2to OHad/Oadis that when switching from O2free solution to O2containing solution,the OCP of Pt/0.1 mol/L HClO4interface shifts promptly from 0.9 V to 1.0 V(Fig.2(d))and from the cyclic voltammetric study with relative fast electrode rotation speed(>800 r/min)and slow potential scan rate (<100 mV/s),it is always observed that the j-E curve for ORR in the negative-going potential scan is smaller than that in the positive-going scan(Fig.3).Our systematic studies also reveal that the faster the electrode rotation speed or the slower the potential scan rate,the larger such difference is.Furthermore,from chronoamperometric transients measured during ORR in the kinetic and kinetic-mass transport mix-controlled potential region,it is seen that ORR currents decay with reaction time(Fig.4).Although trace contaminants cannot be fully avoided,the difference of ORR current in the positive-going scan and that in the negative-going potential scan as well as the decay of chronoamperometric transients for ORR do not originate from contaminants.The reasons are the following:in both O2free and O2containing solution,the cyclic voltammograms are well reproducible when continuously scanning in the potential region from 0.05 V to 1.0 V within 2 h,if there is contaminants accumulation,characteristic current peaks for the redox of the contaminants or continuous decay of the current waves for H-UPD and OHadadsorption/desorption should appear[7];poisoning of the surface with the trace contaminants(such as halides)usually are mass transport controlled,hence its effect on the current decay of ORR under potentiostatic conditions should not depend on the applied electrode potential,i.e.,the current at each reaction potential should be simply reduced by a factor(1-θcontaminats). However,the slope for the current decay is maximum at 0.85 V,and it is smaller at 0.9 and 0.8 V(Fig.4),suggesting that the rate for poison formation is potential dependent(or complete or partially kinetic controlled in nature).
Based on the above experiment facts,we think during ORR there is additional accumulation of OHad/Oadat Pt(111)surface from O2dissociation,the poisoning effect of such accumulated OHadspecies leads to smallerORR current in the subsequent negative-going potential scan or at elongated reaction time under potentiostatic conditions in the kinetic and kinetic-mass transport mix controlled potential region.The faster the electrode rotation speed,the faster the rate for OHadbuilt up from O2dissociation,this leads to larger difference in the ORR current between the negative-going and positivegoing potential scan(Fig.3).It should be noticed that the smaller ORR current in the positive-going scan than that for the negative-going ones at electrode rotation speed below 800 r/min is due to the change relative rate of mass transport of O2to the electrode surface and that of ORR kinetics.When scanning into the kinetic controlled region,the slower ORR kinetics will lead to accumulation of O2near electrode surface,which leads to the higher ORR current the mix-controlled region in the subsequent negative-going potential scan.The fast kinetics for the decomposition of O2to OHad/Oadat H2O+OHadcovered Pt(111)is also supported by DFT calculation,which reveals that activation barrier for this process is just 0.48 eV,it is ca.0.35 eV lower than that for O2decomposition at clean Pt[6].And at H2O+OHadcovered Pt(111),the activation energy for the dissociation of O-O bond in OOHadis only 0.37 eV[6].All the above facts support that the kinetics of f i rst few steps for ORR involving O2adsorption and subsequent decomposition of O2,adto OHador Oad(reactions(2)and(3))are faster than the net rate for OHadreduction to H2O[10].
As estimated from Fig.1 and Fig.2,the current density for OHaddesorption are-7.1 and-2.7 mA/cm2at 0.9 and 0.95 V,which are only 4-7 times higher than that for jk,ORR(-1.8 and-0.35 mA/cm2).Since the difference of the rates for both dissociative adsorption of O2to OHadand that for OHadadsorption/desorption are within 10 times,one should not simply take the f i rst step as r.d.s and the last step as under fast equilibrium, this is also supported by the significantly smaller Tafel slope(-78 mV/dec)than the value of-120 mV/dec. Instead,the kinetics of the current transients can be analyzed according to the simplified reaction scheme given below:
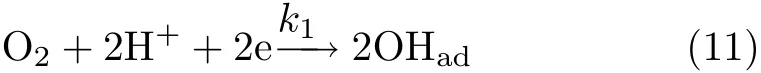
It is followed by reaction(6).
In this scheme,reaction(5)is not only the last step for ORR at Pt in 0.1 mol/L HClO4,but both forward and backward reaction in reaction(6)are also individual reactions which occur fast and in parallel with ORR. Obviously,the dissociative adsorption of O2competes with the dissociation of H2O for the active sites.Ignoring the double layer charging current,the current recorded during linear potential sweeping equals
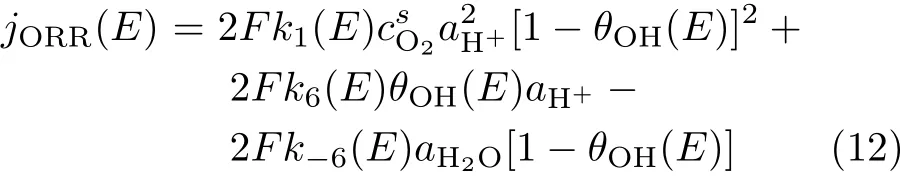
where csO2is the concentration of oxygen near electrode surface.And under potentiostatic condition the ORR current density equals

These equations clearly express that ORR current is controlled by both the rate for OHadformation from O2dissociation and the OHadremoval governed by the reations given reaction(6).This is in contrast to what previously suggested where the f i rst step is r.d.s,and the OHadonly limits free sites available for ORR[2]. It should be mentioned that,a significant difference forof reaction(6)from that of other redox pairs with fi xed ratio of reactants concentration to that of products in bulk solution is thatchanges sensitively with the surface coverage of OHad.From Eq.(7),it is easily seen that when θOHincreases,shifts to positive potentials,and whendecreases,shifts to negative potentials.Hencehas a broad range(i.e.,0.6 V to 1.0 V,Fig.1)rather than just has a single value as usual for other redox pairs dissolved in electrolyte.
If mass transport is fast enough,the fast kinetics of reactions(11)and(6)in both directions soon establish a steady state under which the net rate for O2dissociation to OHadand for OHaddesorption to water equals.It should be mentioned that,for ORR with dissolved O2in electrolyte solution as reactants as usually met in the cases using rotating disk electrode system,the actual current is small since the reaction can easily run into mass transport and kinetic mix controlled mode even with the fastest applicable electrode rotation speed(e.g. 5000 r/min).This renders the applied potential in the kinetic-or mix-controlled potential region for ORR is always just slightly negative of the value forHence, in most RDE studies the net rate for ORR at Pt based electrode is limited by both the thermo-equilibrium of reaction(6)and the slow mass transport for O2to the electrode surface.
IV.CONCLUSION
The kinetics for O2and OHadadsorption/desorption and ORR at Pt(111)electrode in 0.1 mol/L HClO4have been investigated in order to f i nd the key factors which limit ORR kinetics at Pt.It was found that:(i)Eat Pt(111)has a broad range from 0.6 V to 1.0 V,its upper limit determines the onset potential for ORR at Pt(111).(ii)The kinetics for both OHad/Oadformation from O2dissociation and for OHadadsorption from water dissociation and its desorption are very fast,a quasi-steady state will be established very fast.(iii)In the potential range from 0.8 V to 1.0 V,the small deviation of the applied potential toas well as the slow mass transport of O2to the electrode surface renders the net ORR kinetics slow,although the kineticsfor the elementary steps in both forward and backward direction are fast.Our study reveal that adjusting the catalysts properties or reaction conditions,which shiftto positive direction and improve the mass transport of O2,should be key to improve ORR activity for Pt based electrocatalysts.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.20773116),the National Instrumentation Program(No.2011YQ03012416),and 973 Program from the Ministry of Science and Technology of China(No.2010CB923302).
[1]Y.Xu,M.H.Shao,M.Mavrikakis,and R.R.Adzic,in Fuel Cell Catalysis:A Surface Science Approach,New York:Wiley&Sons,9(2009).
[2]S.Gottesfeld,in Fuel Cell Catalysis:A Surface Science Approach,New York:Wiley&Sons,1(2009).
[3]B.B.Blizanac,P.N.Ross,and N.M.Markovic,Electrochim.Acta 52,2264(2007).
[4]F.Tian and A.B.Anderson,J.Phys.Chem.C 115, 4076(2011).
[5]M.Wakisaka,H.Suzuki,S.Mitsui,H.Uchida,and M. Watanabe,J.Phys.Chem.C 112,2750(2008).
[6]V.Tripkovic,E.Skulason,S.Siahrostami,J.K. Norskov,and J.Rossmeisl,Electrochim.Acta 55,7975 (2010).
[7]M.F.Li,L.W.Liao,D.F.Yuan,D.Mei,and Y.X. Chen,Electrochim.Acta 110,780(2013),
[8]M.D.Macia,J.M.Campina,E.Herrero,and J.M. Feliu,J.Electroanal.Chem.564,141(2004).
[9]M.T.M.Koper,Chem.Sci.4,2710(2013).
[10]J.X.Wang,J.Zhang,R.R.Adzic,and J.Phys.Chem. A 111,12702(2007).
[11]N.M.Markovic,T.J.Schmidt,B.N.Grgur,H.A. Gasteiger,R.J.Behm,and P.N.Ross,J.Phys.Chem. B 103,8568(1999).
[12]N.M.Markovic and P.N.Ross,Surf.Sci.Rep.45,121 (2002).
[13]M.H.Shao,P.Liu,and R.R.Adzic,J.Am.Chem. Soc.128,7408(2006).
[14]Y.X.Chen,M.Heinen,Z.Jusys,and R.J.Behm, Angewa.Chem.Int.Ed.45,981(2006).
[15]N.Garcia-Araez,V.Climent,and J.M.Feliu,J.Electroanal.Chem.649,69(2010).
[16]D.M.Drazic,A.V.Tripkovic,K.D.Popovic,and J. D.Lovic,J.Electroanal.Chem.466,155(1999).
[17]K.J.P.Schouten,M.J.T.C.van der Niet,and M.T. M.Koper,Phys.Chem.Chem.Phys.12,15217(2010).
[18]A.S.Bondarenko,I.E.L.Stephens,H.A.Hansen,F.J. Pe′erez-Alonso,V.Tripkovic,T.P.Johansson,J.Rossmeisl,J.K.N?rskov,and I.Chorkendorf f,Langmuir 27,2058(2011).
[19]Q.Mao and U.Krewer,Electrochim Acta 103,188 (2013).
ceived on February 11,2014;Accepted on May 16,2014)
?Author to whom correspondence should be addressed.E-mail:yachen@ustc.edu.cn Tel./FAX:+86-551-63600035
 CHINESE JOURNAL OF CHEMICAL PHYSICS2014年4期
CHINESE JOURNAL OF CHEMICAL PHYSICS2014年4期
- CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Exchange Bias Effect in Phase Separated La0.33Pr0.34Ca0.33MnO3Thin Films
- Elasticity and Thermodynamic Properties of EuS Related to Phase Transition
- Corrosion Study on Tantalum in Anhydrous Ethanol
- Phase Transition Behaviour of VO2Nanorods
- Effect of Molybdenum Doping on Oxygen Permeation Properties and Chemical Stability of SrCo0.8Fe0.2O3-δ
- Preparation of TiO2/Bi2O3Microf i bers and Their Photocatalytic Activity