
Performance Comparison of Two Newly Developed Bimetallic (X-Mo/Al2O3, X=Fe or Co) Catalysts for Reverse Water Gas Shift Reaction
2016-03-22 05:16:38AbolfazlGharibiKharajiAhmadShariati
中國(guó)煉油與石油化工 2016年1期
Abolfazl Gharibi Kharaji; Ahmad Shariati
(1. University of Isfahan, Isfahan, Iran; 2. Petroleum University of Technology, Ahwaz, Iran)
Performance Comparison of Two Newly Developed Bimetallic (X-Mo/Al2O3, X=Fe or Co) Catalysts for Reverse Water Gas Shift Reaction
Abolfazl Gharibi Kharaji1; Ahmad Shariati2
(1. University of Isfahan, Isfahan, Iran; 2. Petroleum University of Technology, Ahwaz, Iran)
The performance of the two newly developed bimetallic catalysts based on the precursor, Mo/Al2O3, was compared for reverse water gas shift (RWGS) reaction. The structures of the precursor and the catalysts were studied using X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET) analysis, inductively coupled plasma-atomic emission spectrometry (ICP-AES), CO chemisorption, temperature programmed reduction of hydrogen (H2-TPR) and scanning electron microscopy (SEM) techniques. The activity of Fe-Mo and Co-Mo catalysts was compared in a fi xed bed reactor at different temperatures. It is shown that the Co-Mo catalyst has higher CO2conversion at all temperature level. The time-on-stream (TOS) analysis of the activity of catalysts for the RWGS reaction was carried out over a continuous period of 60 h for both catalysts. The Fe-Mo/Al2O3catalyst exhibits good stability within a period of 60 h, however, the Co-Mo/Al2O3is gradually deactivated after 50 h of reaction time. Existence of Fe2(MoO4)3phase in Fe-Mo/Al2O3catalyst makes this catalyst more stable for RWGS reaction.
RWGS reaction, bimetallic catalysts, activity, stability.
1 Introduction
Hydrogenation of carbon dioxide has been considered as one of the most economical and effective ways to chemically fix huge amount of emitted carbon dioxide. This process has been attracting more attention for production of hydrocarbons, olefins, methanol and carbon monoxide from carbon dioxide[1-2]. Hydrocarbons production has some shortcomings, including the consumption of a large amount of hydrogen in the reaction and dif fi culties in transporting and storing the product. The synthesis of methanol is dif fi cult because the conversion rate for the direct reaction between carbon dioxide and hydrogen is low. In contrast, the Reverse Water Gas Shift (RWGS) reaction (CO production) has many advantages, including a low hydrogen consumption, a high equilibrium conversion rate and the fact that the CO product can be used as the main raw material for production of major chemical feedstocks.
In recent years, most of the authors[3-8]considered carbon dioxide to be the direct source of methanol production but the yield of methanol produced from CO2is lower than CO[9-10]. The RWGS process can produce CO from CO2; therefore, the yield of methanol production increases when RWGS reaction is used instead of methanol synthesis. This process is named the CAMERE process (carbon dioxide hydrogenation to form methanol via a reversewater-gas shift reaction)[11-12]. The CAMERE process is advantageous for methanol production due to the low costs of the equipment involved and the ease in operation[11].
Carbon dioxide is a highly stable molecule and it does not participate in reaction as an active component. Therefore, CO2reactions need huge activation energy or very effective catalyst. Several studies investigated the RWGS process using one metal oxide as the catalyst on a support. As referred to in the published technical literature, early research work on catalysts was focused on using one metal as the active phase.
In the study by Joo, et al.[13]at a temperature range of 673—973 K, it is shown that the addition of ZnO toAl2O3support would enhance the activity of the catalyst including an increase in its deactivation rate. The deactivation is caused by conversion of zinc oxide molecules to zinc ions. Copper-based catalysts are not suitable for operating at high temperature because of its poor thermal stability. By adding a small amount of iron in the study conducted by Chen, et al.[14]the catalytic activity and stability of copper-based catalysts at high temperature were effectively improved. In the study conducted by Wang, et al.[15]the Ni-CeO2catalysts with different Ni contents were prepared by a co-precipitation method and used for RWGS reaction. 2% of Ni on CeO2showed excellent catalytic performance in terms of its activity, selectivity, and stability for RWGS reaction. In the study performed by Kim, et al.[16]the effect of the morphological characteristics of a TiO2support on the reverse water gas shift reaction over Pt/TiO2catalysts was examined. The results indicated that the active sites of the Pt/TiO2catalyst included both the Pt sites and the TiO2sites, which were reducible, and the difference in the activity of the Pt/TiO2catalysts was dependent on the reducibility of the TiO2supports. Since the TiO2crystallite size was a key factor dictating the reducibility of the TiO2sites, they concluded that the catalytic activity of Pt/TiO2in the reverse water gas shift reaction was affected to a greater extent by the properties of the TiO2supports than by the structure of the Pt. Progress has been made as stated above, however, the stable and selective catalysts operating at high temperature for RWGS reaction are still in a developing stage. The results of experiments performed by Goguet, et al.[17]highlight the potential role played by carbon deposition in the deactivation of the 2% Pt/CeO2catalyst under RWGS reaction conditions. The results obtained thereby seemed to indicate that the carbon deposition is located primarily on the support (ceria) and not on the platinum. This result indicates that the support was “active” in the reaction and that the deactivation took place through gradual carbon coverage of the support.
The combination of two metals in an oxide matrix can produce materials with novel physical and chemical properties. In these catalysts a metal plays the role of active site, with another functioning as the promoter and/or distributor. For example, in the study made by Chen, et al.[18]on Cu/K2O/SiO2catalyst, copper is the active site and potassium plays the role of promoter. In this study the reverse water gas shift (RWGS) reaction over Cu/SiO2with and without potassium promoter was studied. After addition of even a little amount of potassium, the Cu/ K2O/SiO2catalyst obviously offers better catalytic activity than the Cu/SiO2catalyst. The main role of K2O was to provide catalytic activity for decomposition of formates, besides acting as a promoter for CO2adsorption. Just very recently Gharibi Kharaji, et al.[19]developed the Ni-Mo/Al2O3catalyst for RWGS reaction, and in this study Mo had the dispersion and promoter roles. The existence of Mo can generate an electron de fi cient state on the Ni species in NiMoO4phase of catalyst. In another study, Gharibi Kharaji, et al.[20]examined the performance of a novel bimetallic Fe-Mo/Al2O3catalyst for RWGS reaction. The activity of this catalyst was nearly tantamount to an equilibrium conversion in a batch reactor at 600 ℃. This catalyst was stable for 60 h in a fi xed bed reactor at a temperature of 600 ℃ and a GHSV of 30 000 mL/(h·g) using an 1:1 molar ratio of feed. The results indicated that Mo existence in the structure of the Fe-Mo/Al2O3catalyst enhances its activity as compared to the Fe/Al2O3catalyst. This enhancement is attributed to better Fe dispersion and smaller particle size of Fe species. Gharibi Kharaji and Shariati[8]introduced another bimetallic Co-Mo/Al2O3catalyst for RWGS reaction. The activity of this catalyst was also close to the equilibrium conversion in a batch reactor at 600 ℃ for a 1:1 molar ratio feed. The time on stream test of stability was not performed for this catalyst, however.
To be scaled up for industrial use, more rigorous process tests on bimetallic catalysts are needed in a fl ow reactor under harsh operating conditions. Therefore, this study intends to compare the performance of the two newly developed bimetallic catalysts (X-Mo/Al2O3, X= Fe or Co) under the same harsh operating conditions in a fi xed bed reactor. To describe the behavior of these catalysts in the process tests, more explicit characterization tests were also performed.
2 Experimental
2.1 Catalyst preparation
3.2 g of molybdenum complex (NH4)6Mo7O24·4H2O(Merck) was dissolved in 200 mL of distilled water, and then this solution was added to a slurry of alumina consisting of 10 g of γ-Al2O3(Merck Co., 170 m2/g, 99% pure, porosity=1.3 cm3/g, pore diameter=18 nm) and 300 mL of distilled water. The solution was stirred by a high speed mechanical stirrer for 10 h at 308 K. In this process, molybdate anions were chemisorbed on the surface of γ-Al2O3particles, so that the Mo/Al2O3serving as a precursor was obtained. The Fe-Mo/Al2O3and Co-Mo/ Al2O3catalysts were synthesized by slowly adding an iron salt solution (4.4 g of Fe(NO3)2·6H2O (Merck Co., 99% pure) in 200 mL of distilled water) and a cobalt salt solution (4.4 g of Co(NO3)2·6H2O (Merck Co.), 99% pure) in 200 mL of distilled water) to the precursor, the Mo/Al2O3slurry (10 g in 300 mL of distilled water), respectively. The stirring conditions for both catalysts were similar, and after that procedure the impregnated samples were subsequently air-dried at 323 K for 10 h and then were calcined in air at 923 K for 5 h.
2.2 Catalyst characterizations
The structures of the precursor and catalysts were studied by X-ray diffraction analysis conducted on a PW1840 X-ray powder diffractometer (Phillips, Netherland) using CuKαradiation operated at a tube voltage of 40 kV and a tube current of 30 mA with a step size of 0.02° and a scanning angle ranging from 5° to 90°.
The speci fi c surface areas of the samples were determined using the Brunauer–Emmett–Teller method with adsorption of nitrogen at liquid nitrogen temperature followed by subsequent desorption at room temperature after initial pretreatment of the samples by degassing at 573 K for 1 h. The BET surface area was obtained with a Quantasorb surface area analyzer (Quantachrome, USA). Temperature programmed reduction was conducted on a U-shaped quartz tube embedded in a programmable furnace. 80 mg of the catalyst were pre-treated with pure He flow at 573 K for 1 h and then were reduced under a gas mixture fl ow (5% of H2and 95% of Ar at a fl ow rate of 40 mL/min). The TPR patterns were obtained by using a recorder connected to a GC equipped with a TCD in a temperature range of 430—1 250 K at a heating rate of 10 K/min. The metal (cobalt and iron) particle size of the reduced catalysts was determined by irreversible CO adsorption at 303 K by BELCAT-B chemisorption analyzer (BEL Inc., Japan). The CO uptake was measured by injecting successively 0.5 mL of CO samples via a calibrated loop into 25 mL/min of He carrier until a constant peak area was reached. The chemical composition of bulk medium was determined with an ICP-AES emission spectroscope (Perkin Elmer, Optima 2000DV).
The energy dispersive X-ray spectroscopy (EDX) used a scanning electron microscope (SEM) equipped with an energy dispersive X-ray (EDX) analyzer at a voltage of 17 kV and an acquisition lifetime of 160 s for analyzing the chemical composition of the catalysts. Scanning electron microscopy (SEM) images were recorded with an XL300 SEM (Philips, Netherland). The samples were coated with gold before the measurement. In this study, SEM tests were carried out between a magnification of 100 to 40 000 times, but for the sake of better comparison the micrographs with a magnification of 20 000 times were chosen.
2.3 RWGS reactor system
The catalytic performance tests for the two catalyst samples were conducted in a fi xed-bed reactor. A schematic diagram of the experimental apparatus and the con fi guration of the reactor are shown in Figure 1. The reaction tube (20 mm in ID and 150 mm in length) was made from stainless steel. The mass flow rate was controlled and measured using mass fl ow controllers (Hitachi). The stability of the catalysts was investigated under the reaction conditions covering a temperature of 873 K and a GHSV of 80 000 (mL/h·g) with a molar ratio of 1 under atmospheric pressure. Prior to the reaction, the catalysts were reduced in situ at 923 K for 4 h under a 200 mL/min fl ow of hydrogen and nitrogen mixture with a hydrogen to nitrogen ratio of 1/5. A cold trap at the outlet of the reactor was used to condense out any water from the gas product stream.
All products were analyzed by a gas chromatograph (Young Lin) equipped with Q and MS capillary columns and a HID detector. CO, H2,CO2, and CH4were detected by GC and their respective mole fractions were calculated from peak area with a third order calibration function. The initial CO2conversion experiment was repeated fi ve times for each of the catalysts. The test results led to anestimated accuracy of ±3% in experimental measurements.
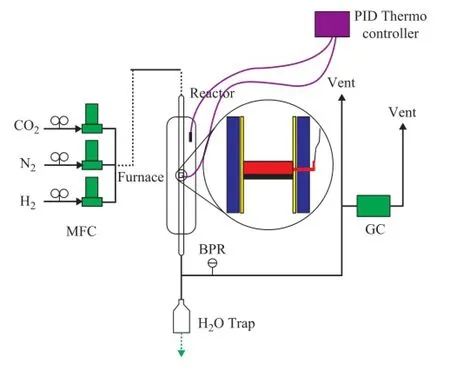
Figure 1 A schematic diagram of the fi xed-bed reactor
3 Results and Discussion
Figure 2 shows the X-ray diffraction patterns of the Mo/ Al2O3, Co-Mo/Al2O3and Fe-Mo/Al2O3catalysts. The phases Al2O3(PDF 073-1512) and MoO3(PDF 01-0706) were recognized in the XRD patterns of all catalysts. The phases Al2(MoO4)3(PDF 023-0764) and Fe2(MoO4)3(PDF 035-0183) were identi fi ed in the X-ray diffraction patterns of the Fe-Mo/Al2O3catalyst. Fe2(MoO4)3is consistent with the suggestion that Fe2O3and MoO3can easily form Fe2(MoO4)3after heat treatment[20]. The phases CoMoO4(PDF 021-0868) and Al2(MoO4)3(PDF 023-0764) were identi fi ed in the X-ray diffraction patterns of the Co-Mo/Al2O3catalyst. The XRD results indicate that the CoMoO4phase was apparent, when cobalt was added to Mo/Al2O3. The existence of CoMoO4phase in the Co-Mo/Al2O3catalyst after calcination shows that this phase was formed by the solid state reaction between Co2O3and MoO3.
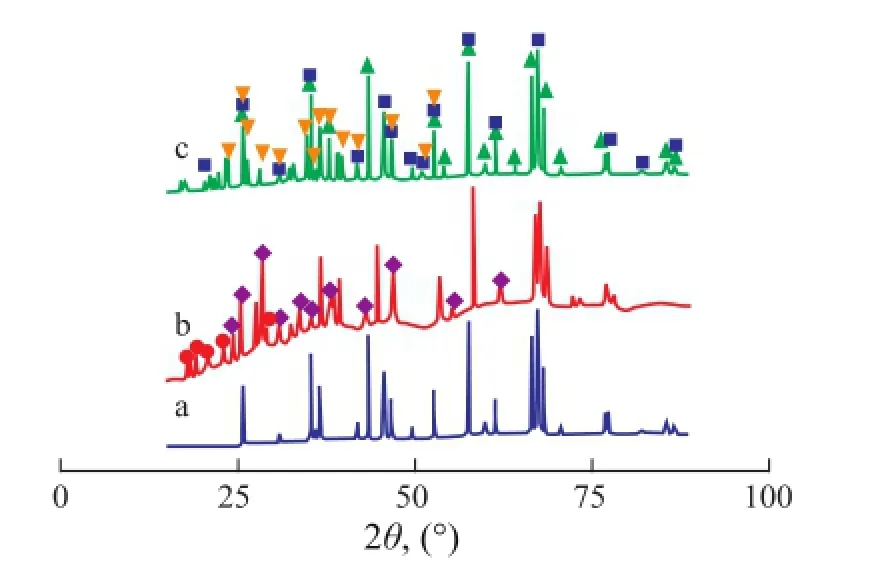
Figure 2 The XRD patterns for calcined Mo/Al2O3(a), Co-Mo/Al2O3(b) and Fe-Mo/Al2O3(c) catalysts
The metal concentration, average pore diameter, BET surface area and metal particle size of the precursor and catalysts are shown in Table 1. Iron and cobalt promoter incorporation decreased total surface area of the catalysts. In our previous work[20]Fe species in the Fe-Mo/Al2O3catalyst had higher extent of dispersion than the Fe/Al2O3catalyst, so Mo existence in the structure of Fe-Mo/Al2O3increased the Fe dispersion and at the same time the speci fi c surface area of the catalyst did not change. Table 1 con fi rms the same results for both catalysts in this study. Therefore it can be concluded that Mo existence in the Fe-Mo/Al2O3and the Co-Mo/Al2O3catalysts has similar dispersion effect.
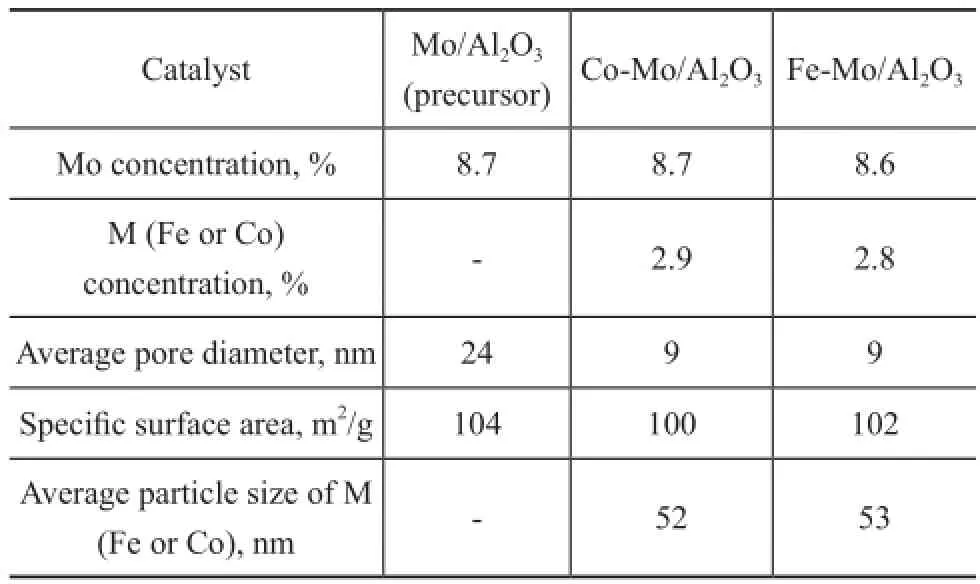
Table 1 Specific surface area and weight percent of Fe and Mo species for each catalyst
The H2-TPR pro fi les of the catalysts are shown in Figure 3. During the H2-TPR tests, no H2consumption was observed in any case below 500 K. According to our pervious study[20], the TPR patterns of the Mo/Al2O3catalyst showed two peaks found approximately at 765 K and 990 K, The fi rst peak was ascribed to polymeric Mo species which corresponded to Mo4O11formed during reduction of MoO3, and the second peak was attributed to the reduction of Mo4O11to form MoO2.
It is clear that Mo incorporation signi fi cantly affects the reduction behaviour of the bimetallic iron and cobalt catalysts. Mo existence in the structure of Fe-Mo/Al2O3catalyst adds a peak at the higher temperature side of thesecond peak. According to the XRD results, this peak (the third peak) could be attributed to the reduction-resistant Fe–Mo composite oxides (possibly existing as the Fe2(MoO4)3phase). The reduction of the Co-Mo/Al2O3catalyst begins at about 560 K and proceeds in two essential steps observed in the temperature ranges of 685—840 K and 840—1 135 K, respectively. The low temperature effect with a relatively low intensity peak corresponds to the reduction of Mo–O and Co–O species which are weakly bonded to the alumina support, whereas at a medium temperature, one of the peaks can be attributed to the reduction of Mo6+to Mo4+. At temperatures above 840 K a deep reduction of Mo4+to Mo0and Co3+to Co0would occur[13]. In this temperature range, a major reduction peak for transforming Co2O3to Co overlaps the reduction peak of[20].
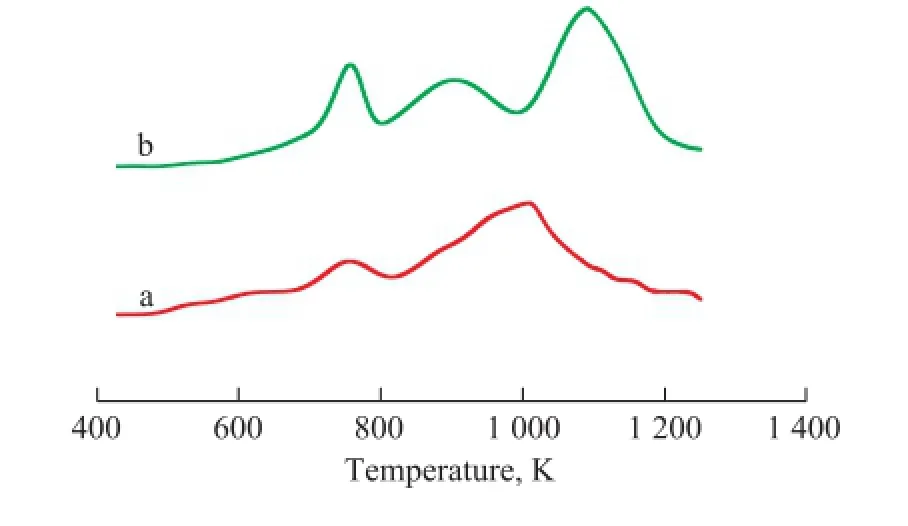
Figure 3 he H2-TPR patterns for calcined Co-Mo/Al2O3(a) and Fe-Mo/Al2O3(b) catalysts
Figure 4 illustrates the dependence of CO2conversion on the mole fraction of H2in the H2/CO2feed. With an increasing H2concentration, CO2conversion over the Fe-Mo and Co-Mo catalysts passes over a volcano showing a maximum value at H2/CO2= 1, while a similar phenomenon was reported by Chen, et al.[21]for the Cu/Al2O3catalyst. The CO2conversion over the Co-Mo catalyst also reached a maximum when H2in the feed gas accounted for 50 mole %. However, the slope showing the decrease in conversion after that point was lower than the case of the Fe-Mo catalyst. Therefore, the performance of both catalysts was investigated at a H2to CO2molar ratio of 1 afterwards.
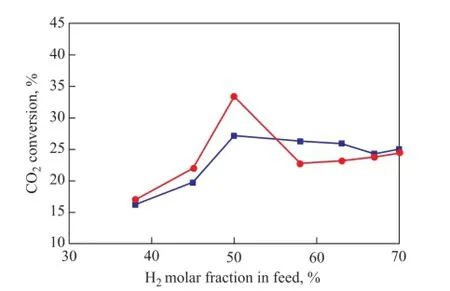
Figure 4 CO2conversion versus mole percent of H2in feed for both catalysts (reaction condition: 873 K and GHSV= 50 000 mL/h·g)
The activity of Fe-Mo and Co-Mo catalysts was compared in a fixed bed reactor, as shown in Figure 5. The same feed composition (CO2/H2=1) and space velocity (GHSV= 50 000 mL/h·g) were used for both catalysts. The reactivity of both catalysts for RWGS reaction was investigated at different reaction temperatures. The conversion of CO2increased with the reaction temperature as it increased from 673 K to 973 K. It can be seen that the Co-Mo catalyst has higher CO2conversion at all temperature range. Based on these results and the XRD analyses, it is clear that the CoMoO4phase in the Co-Mo catalyst was more active than Fe2(MoO4)3phase in the Fe-Mo catalyst for RWGS reaction.
In the literature [21-22], the redox and formate decomposition mechanisms were proposed to explain the mechanism of CO formation in the RWGS reaction. According to this model when the surface reduction is easily done, the CO formation and also CO2conversion are enhanced. Therefore the Co-Mo catalyst, which is characteristicof higher surface reducibility than the Fe-Mo catalyst, achieves higher CO2conversion.
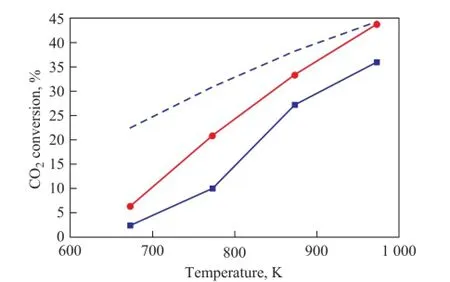
Figure 5 The CO2conversion versus temperature for both catalysts (at CO2/H2= 1:1, and GHSV= 50 000 mL/h·g)
The time-on-stream (TOS) analysis of the catalytic activity for the RWGS reaction was carried out over a continuous period of 60 h for both catalysts at a temperature of 873 K and a GHSV of 80 000 mL/h·g. These results are shown in Figure 6. A high feed fl ow was selected to eliminate the thermodynamic limitation of RWGS reaction and to verify the stability of catalyst in a short time. It is evident that the Fe-Mo/Al2O3catalyst exhibits good stability within a period of 60 h, however, the Co-Mo/ Al2O3is gradually deactivated after 50 h of reaction time.
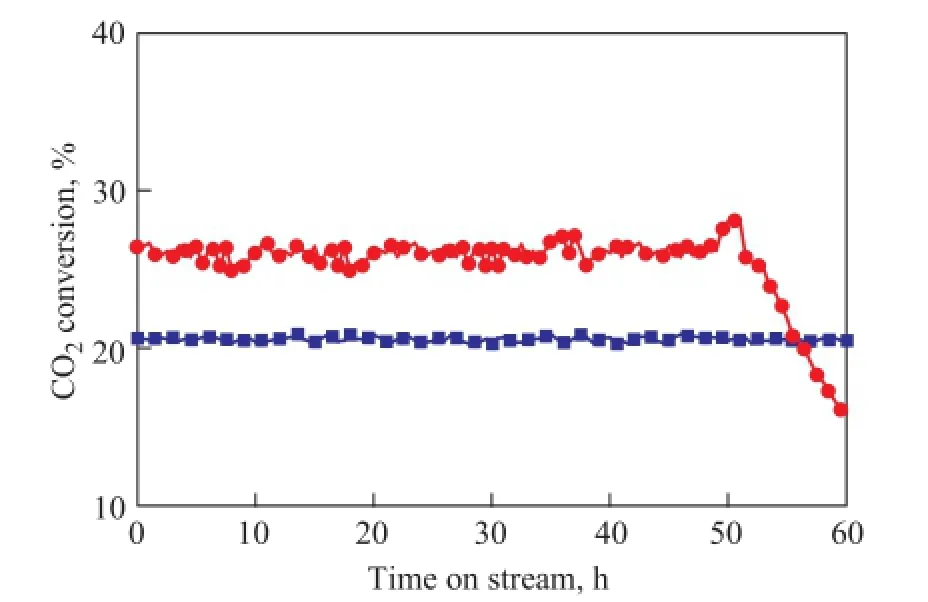
Figure 6 Stability of both catalysts in RWGS reaction with respect to time on stream (reaction condition: at 873 K, CO2/ H2=1:1 and GHSV= 80 000 mL/h·g)
The SEM analysis was performed on the fresh catalysts (after reduction) and the used catalysts (after 60 h on stream at a temperature of 873 K and a GHSV of 80 000 mL/h·g). This analysis was conducted for the used catalysts to obtain a deeper understanding on their morphology and structure. The SEM images of the fresh and used Fe-Mo/Al2O3catalysts are given in Figures 7 and 8, respectively. Since the molecular weight of Mo is higher than Fe, its metallic form should be brighter in SEM micrographs, and consequently the brighter Mo species are expected if they are present in their reduced metallic form. The XRD patterns confirm that Mo species is present in its oxidized form (MoO3), therefore it is not observed as bright spots in SEM images. It is concluded that the bright spots observed on the particles in Figure 7 are Fe crystallites that are irregularly spread on the surface. The Fe-Mo/ Al2O3catalyst exhibits an agglomerated/mixed structure which indicates that the Fe crystallites are located either on the top or close to MoO3species that were previously impregnated on the support surface. It is well known that the type and amount of carbon deposits are crucial factors that affect the activity and stability of the catalysts. Figure 8 does not show any carbon deposits formed during reaction on the used Fe-Mo/Al2O3catalyst surface.

Figure 7 SEM image of fresh Fe-Mo/Al2O3catalyst (×20000)
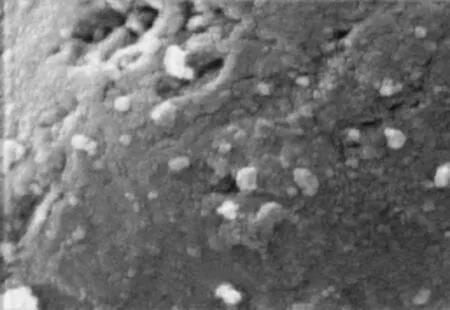
Figure 8 SEM image of used Fe-Mo/Al2O3catalyst (×20000)
The SEM images of the fresh and used Co-Mo/Al2O3catalyst are given in Figures 9 and 10, respectively. The bright spots observed on the particles in Figure 9 are the cobalt crystallites that are irregularly spread on the surface. As it is mentioned, the type and amount of carbon deposits are crucial factors that affect the activity and stability of the catalysts. Figure 10 shows carbon deposition on the used catalyst surface in the form of carbon fi bers during the reaction. These carbon fi bers occupy the active sites and pore volume of catalyst and are responsible for catalyst deactivation. The EDX analysis shows that carbon composition in the surface of used Co-Mo/Al2O3and Fe-Mo/Al2O3catalysts was 26% and 1%, respectively. According to the analytical results, the active sites of Co-Mo catalyst are more active than the Fe-Mo catalyst. Therefore the dissociation of CO2to CO and coke formation is further studied on the CO-Mo catalyst.

Figure 9 SEM image of the fresh Co-Mo/Al2O3catalyst (×20000)

Figure 10 SEM image of the used Co-Mo/Al2O3catalyst (×20000)
The XRD patterns of fresh and used Fe-Mo/Al2O3and Co-Mo/Al2O3catalysts are illustrated in Figures 11 and 12, respectively. The location of Fe2(MoO4)3and CoMoO4phases in XRD patterns where they do not overlap other solid phases are shown in these Figures. Fe2(MoO4)3was observed on fresh and used Fe-Mo/Al2O3catalysts, but most of the peaks of CoMoO4vanished in the XRD patterns of used Co-Mo/Al2O3catalyst. Therefore, it is concluded that Fe2(MoO4)3phase remained in the structure of Fe-Mo/Al2O3catalyst, in contrast to the CoMoO4phase in the Co-Mo/Al2O3catalyst during RWGS reaction. The XRD analysis results also indicate that the Fe–O–Mo structure could crystallize to form the Fe2(MoO4)3phase. Fe2(MoO4)3is monoclinic, with a structure containing isolated MoO4tetrahedrons and FeO6octahedrons that share corners, in which each oxygen atom is bonded only to one Fe atom and one Mo atom[20]. According to the H2-TPR results, Fe2(MoO4)3is not a easily reducible phase, since it has a higher reduction temperature than iron oxides. It is conceivable that the Fe–O–Mo structure would inhibit the reduction of catalysts[23].
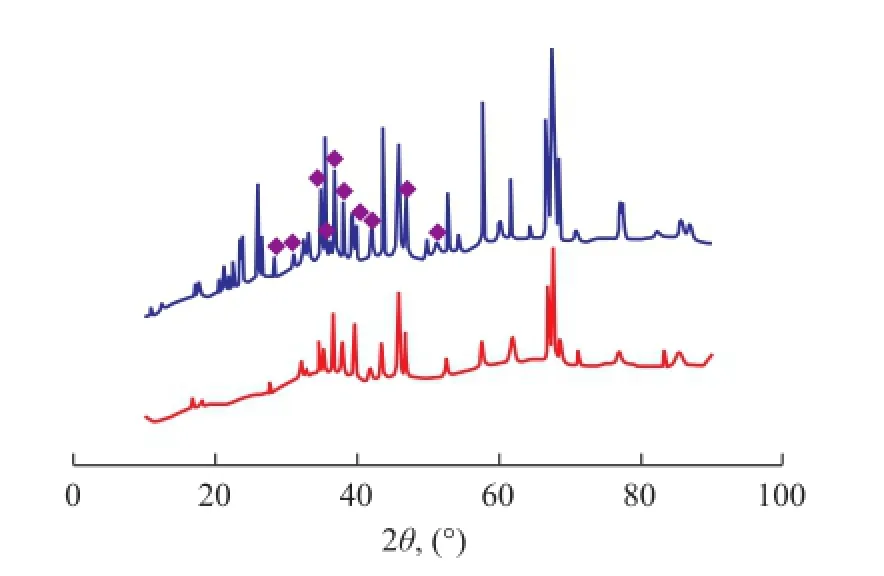
Figure 11 XRD patterns of fresh and used Fe-Mo/Al2O3catalysts

Figure 12 XRD patterns of fresh and used Co-Mo/Al2O3catalysts
4 Conclusions
Since the CoMoO4and Fe2(MoO4)3phases are responsible for the activity of Co-Mo/Al2O3and Fe-Mo/Al2O3catalysts, respectively, it appears that the CoMoO4phase is more active than the Fe2(MoO4)3phase. However, the Co-MoO4phase is more prone to deactivation due to the surface carbon formation. These carbon fi bers occupy active sites and pore volume of catalyst and are responsible for catalyst deactivation. The existence of Fe2(MoO4)3phase in the Fe-Mo/Al2O3catalyst make this catalyst more stable during RWGS reaction. The Fe2(MoO4)3phase in the Fe-Mo/Al2O3catalyst shows low reducibility and remains in structure through 60 h of reaction time.
Acknowledgements: Authors gratefully acknowledge the Iranian Nano Technology Initiative Council and the Petroleum University of Technology for fi nancial support.
[1] Song C. Global challenges and strategies for control, conversion and utilization of CO2for sustainable development involving energy, catalysis, adsorption and chemical processing[J]. Catalysis Today, 2006, 115(1/4): 2-32
[2] Kim S S, Lee H H, Hong S C. A study on the effect of support’s reducibility on the reverse water-gas shift reaction over Pt catalysts[J]. Applied Catalysis A: General, 2012, 423/424: 100-107
[3] Ostrovskii V E. Mechanisms of methanol synthesis from hydrogen and carbon oxides at Cu–Zn-containing catalysts in the context of some fundamental problems of heterogeneous catalysis[J]. Catalysis Today, 2002, 77(3): 141-160
[4] Bussche K M V, Froment G F. A steady-state kinetic model for methanol synthesis and the water gas shift reaction on a commercial Cu/ZnO/Al2O3catalyst[J]. Journal of Catalysis, 1996, 161(1): 1-10
[5] Sahibzada M, Metcalfe I S, Chadwick D. Methanol synthesis from CO/CO2/H2over Cu/ZnO/Al2O3at differential and finite conversions[J]. Journal of Catalysis, 1998, 174(2): 111-118
[6] Sun Q, Zhang Y L, Chen H Y, et al. A novel process for the preparation of Cu/ZnO and Cu/ZnO/Al2O3ultra fi ne catalyst: Structure, surface properties, and activity for methanol synthesis from CO2+H2[J]. Journal of Catalysis, 1997, 167(1): 92-105
[7] Gharibi Kharaji A,. Shariati A, Takassi M A. Selectivity and performance of Fe-V2O5/γ-Al2O3nano catalyst for methanol production with reverse water gas shift (RWGS) reaction[J]. Journal of American Science, 2011, 7(12): 1054-1068
[8] Gharibi Kharaji A, Shariati A. Performance of Co-Mo/Al2O3nano catalyst for CAMERE process in a batch reactor[J]. Journal of Chemical and Biochemical Engineering Quarterly, 2013, 27(3): 275-278
[9] Edwards J H. Potential sources of CO2and the options for its large-scale utilisation now and in the future[J]. Catalysis Today, 1995, 23(1): 59-66
[10] Gharibi Kharaji A, Shariati A. Selectivity and performance of Ni-Mo/γ-Al2O3catalyst for methanol production with reverse water gas shift (RWGS) reaction[J]. Journal of American Science, 2012, 8(8): 265-270
[11] Joo O S, Jung K D, Moon I, et al. Carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction (the CAMERE process) [J]. Industrial & Engineering Chemistry Research, 1999, 38 (5): 1808-1812
[12] Gharibi Kharaji A, Shariati A, Takassi M A. Novel catalysts for carbon dioxide utilization[M]. Germany: LAP LAMBERT Academic Publishing, 2013
[13] Joo O S, Jung K D. Stability of ZnAl2O4catalyst for reverse-water-gas-shift reaction (RWGSR) [J]. Bulletin of the Korean Chemical Society, 2003, 24(1): 86-90
[14] Chen C S, Cheng W H, Lin S S. Study of iron-promoted Cu/SiO2catalyst on high temperature reverse water gas shift reaction[J]. Applied Catalysis A: General, 2004, 257(1): 97-106
[15] Wang L, Zhang S, Liu Y. Reverse water gas shift reaction over Co-precipitated Ni-CeO2catalysts[J]. Journal of Rare Earths, 2008, 26(1): 66-70
[16] Kim S S, Lee H H, Hong S C. The effect of the morphological characteristics of TiO2supports on the reverse water–gas shift reaction over Pt/TiO2catalysts[J]. Applied Catalysis B: Environmental, 2012, 119/120: 100-108
[17] Goguet A, Meunier F, Breen J P, et al. Study of the origin of the deactivation of a Pt/CeO2catalyst during reverse water gas shift (RWGS) reaction[J]. Journal of Catalysis, 2004, 226(2): 382-392
[18] Chen C S, Cheng W H, Lin S S. Study of reverse water gas shift reaction by TPD, TPR and CO2hydrogenation over potassium-promoted Cu/SiO2catalyst[J]. Applied Catalysis A: General, 2003, 238: 55-67
[19] Gharibi Kharaji A, Shariati A, Ostadi M. Development of Ni-Mo/Al2O3catalyst for reverse water gas shift (RWGS) reaction[J]. Journal of Nanoscience and Nanotechnology, 2014, 14(9): 6841-6847
[20] Gharibi Kharaji A, Shariati A, Takassi M A. A novel γ-alumina supported Fe-Mo bimetallic catalyst for reverse water gas shift reaction[J]. Chinese Journal of Chemical Engineering, 2013, 21(9): 1007-1014
[21] Chen C S, Cheng W H, Lin S S. Mechanism of CO formation in reverse water–gas shift reaction over Cu/Al2O3catalyst[J]. Catalysis Letters, 2000, 68(1): 45-48
[22] Ginés M J L, Marchi A J, Apesteguía C R. Kinetic study of the reverse water-gas shift reaction over CuO/ZnO/Al2O3catalysts[J]. Applied Catalysis A: General, 1997, 154(1/2): 155-171
[23] Qin S, Zhang C, Xu J, et al. Fe–Mo interactions and their influence on Fischer–Tropsch synthesis performance[J]. Applied Catalysis A: General, 2011, 392(1/2): 118-126
Received date: 2015-11-13; Accepted date: 2015-12-23.
E-mail of Gharibi Kharaji: abolfazl. gharibi@yahoo.com; E-mail of Shariati: shariati@put.ac.ir.

- 中國(guó)煉油與石油化工的其它文章
- Numerical Study of Air Nozzles on Mild Combustion for Application to Forward Flow Furnace
- Molecular Simulations of FCC Dry Gas Components Adsorption in Zeolite Y1
- Intrinsic Kinetic Modeling of Thermal Dimerization of C5Fraction
- Synthesis and Properties of Dendritic Long-Chain Esters as Crude Oil Flow Improver Additives
- Method for Control of Particle Size and Morphology of Paraf fi n/Polystyrene-Divinylbenzene Microcapsules
- Study on Absorption and Regeneration Performance of Novel Hybrid Solutions for CO2Capture