
Web Resources for Metagenomics Studies
2015-12-21 03:13:16PravinDudhagaraSunilBhavsarbChintanBhagatcAnjanaGhelaniShreyasBhattRajeshPatel
Pravin Dudhagara*,Sunil Bhavsarb,Chintan Bhagatc,Anjana Ghelani, Shreyas Bhatt,Rajesh Patel
1Department of Biosciences,Veer Narmad South Gujarat University,Surat 395007,India
2Department of Microbiology,Shree Ramkrishna Institute of Computer Education and Applied Sciences,Surat 395001,India
3Department of Life Sciences,Hemchandracharya North Gujarat University,Patan 384265,India
RESOURCE REVIEW
Web Resources for Metagenomics Studies
Pravin Dudhagara1,*,a,Sunil Bhavsar1,b,Chintan Bhagat1,c,Anjana Ghelani2,d, Shreyas Bhatt3,e,Rajesh Patel3,f
1Department of Biosciences,Veer Narmad South Gujarat University,Surat 395007,India
2Department of Microbiology,Shree Ramkrishna Institute of Computer Education and Applied Sciences,Surat 395001,India
3Department of Life Sciences,Hemchandracharya North Gujarat University,Patan 384265,India
Available online 18 November 2015
Handled by Vladimir B.Bajic
Metagenomics; Metagenomes; Web resources; Software tools; Synthetic metagenome
The developmentofnext-generation sequencing(NGS)platforms spawned an enormous volume of data.This explosion in data has unearthed new scalability challenges for existing bioinformatics tools.The analysis of metagenomic sequences using bioinformatics pipelines is complicated by the substantial complexity of these data.In this article,we review several commonly-used online tools for metagenomics data analysis with respect to their quality and detail of analysis using simulated metagenomics data.There are at least a dozen such software tools presently available in the public domain.Among them,MGRAST,IMG/M,and METAVIR are the most well-known tools according to the number of citations by peer-reviewed scientific media up to mid-2015.Here,we describe 12 online tools with respect to their web link,annotation pipelines,clustering methods,online user support,and availability of data storage.We have also done the rating for each toolto screen more potentialand preferentialtools and evaluated five best tools using synthetic metagenome.The article comprehensively deals with the contemporary problems and the prospects of metagenomics from a bioinformatics viewpoint.
Introduction
Metagenomics has become an emerging and indispensable field of microbial ecology since the first use of the concept in 1998 [1].It deals with the study of genetic material recovered directly from environmental samples.Originally,metagenomics studies depended on the cultivation of clonalcultures followed by functional expression screening[1],which are not able to represent the total community profile and may overlook the vast majority of the microbial biodiversity [2].The advent of next-generation sequencing(NGS)technologies has made it possible to perform sequencing-basedmetagenomic analyses.NGS platforms allow researchers to retrieve high-throughput data at substantially lower cost,fueling the development of sequence-based metagenomics analyses aimed at decoding the entire microbial community[3].
Two distinctmetagenomics approaches are commonly used: the first is referred to as marker-gene metagenomics or targeted metagenomics,and the second is shotgun metagenomics.The former involves the use of marker genes(including conserved markers such as 16S rRNA[4,5],18S rRNA,and ITS[6])and an amplicon sequencing method to reassemble the taxonomic structure of a microbial community[7,8].For the latter approach,the random nature of shotgun sequencing ensures adequate coverage to assess the entire community’s structure [9,10]and opens new avenues for discovering structural and functional novelties[11].At present,thousands of markergene assisted and shotgun metagenomics projects have been undertaken,comprising millions of samples available in the public domain.After sequencing of a metagenomics sample, the primary task is to analyze the vast amount of data in order to determine the identities and roles ofthe microbialspeciespresentin the sample[12].Severaloffline toolsare available forclassifying metagenomic reads against known reference datasets [13].However,the assembly of metagenomic sequence data and the identification of the operational taxonomic units (OTUs)are major technical challenges that persist in metagenomic data analysis pipelines[14].Moreover,the high complexity of metagenomics data is a criticalbarrier in analyzing these metadata using stand-alone tools.On the other hand,various web-based workflows may be able to offer in-depth exploration without the need for highly configured computers.
In this article,we review the most useful online software tools for metagenomics data analysis,including a brief description of how they work in the course of the analysis. More importantly,we have rated the tools on the basis of overall users’experience in five vital criteria:(1)ease of data uploading,(2)availability of online user support,(3)spectrum of data analyses available,(4)number of citations in scientific literature,and(5)size of the stored data(Figure 1, Table 1).While such an evaluation can never be completely thorough,we nevertheless hope that our evaluation of these web resources for metagenomics studies will provide vital information for budding scientists,assisting them to make the best choice for their preferred applications.
MG-RAST
The software package Metagenomics Rapid Annotation using Subsystem Technology(MG-RAST)is a user-friendly opensource server initially constructed on the SEED project (http://www.theseed.org/)[15]framework for metagenomics studies[16–18].Debuting in 2007,MG-RAST was one of the earliest online metagenomics analysis tools[16].A new version (GitHub embedded version 3.6)is currently available,which does not entirely depend on SEED technology but uses the SEED subsystem instead as a preferred data source,to enable taxonomic and functional classification of metagenomes.The new version is encapsulated and separated from the data store, allowing far greater scalability and much faster computation than previous versions.Currently,MG-RAST contains 215,773 metagenomic datasets,which have been accessed by more than 12,000 registered users.Other than data access,registered users can also submit their own raw metagenomic data in FASTA,FASTQ,and SFF formats along with detailed sample information.The uploaded data are processed by a multi-step workflow that includes quality control,automated annotation,and analysis of prokaryotic metagenomic shotgun samples as well as targeted samples.Automated annotation is done using numerous reference datasets.The server supports a variety of user-driven analyses,including phylogenetic,functional,metabolic,and comparative analyses of two or more metagenomes.MG-RAST also provides the facility to select a database for quantitative insights into microbialpopulations based on sequencing data.It also offers the ability to output data in multiple clustering forms that are downloadable in flattext format or to export data as FASTA and QIIME files. Registered users have the right to publish their data openly or keep data private and can share data among multiple users with protected confidentiality.In these ways,MG-RAST offers great flexibility in analysis,privacy,and sharing of data. Therefore,it is the most reviewed and explored tool in use today[8,13,19].So far,more than 2000 metagenome projects and over 13,000 metagenomic datasets have been uploaded to the MG-RAST servers each month of 2015,indicating the great popularity of the tool.
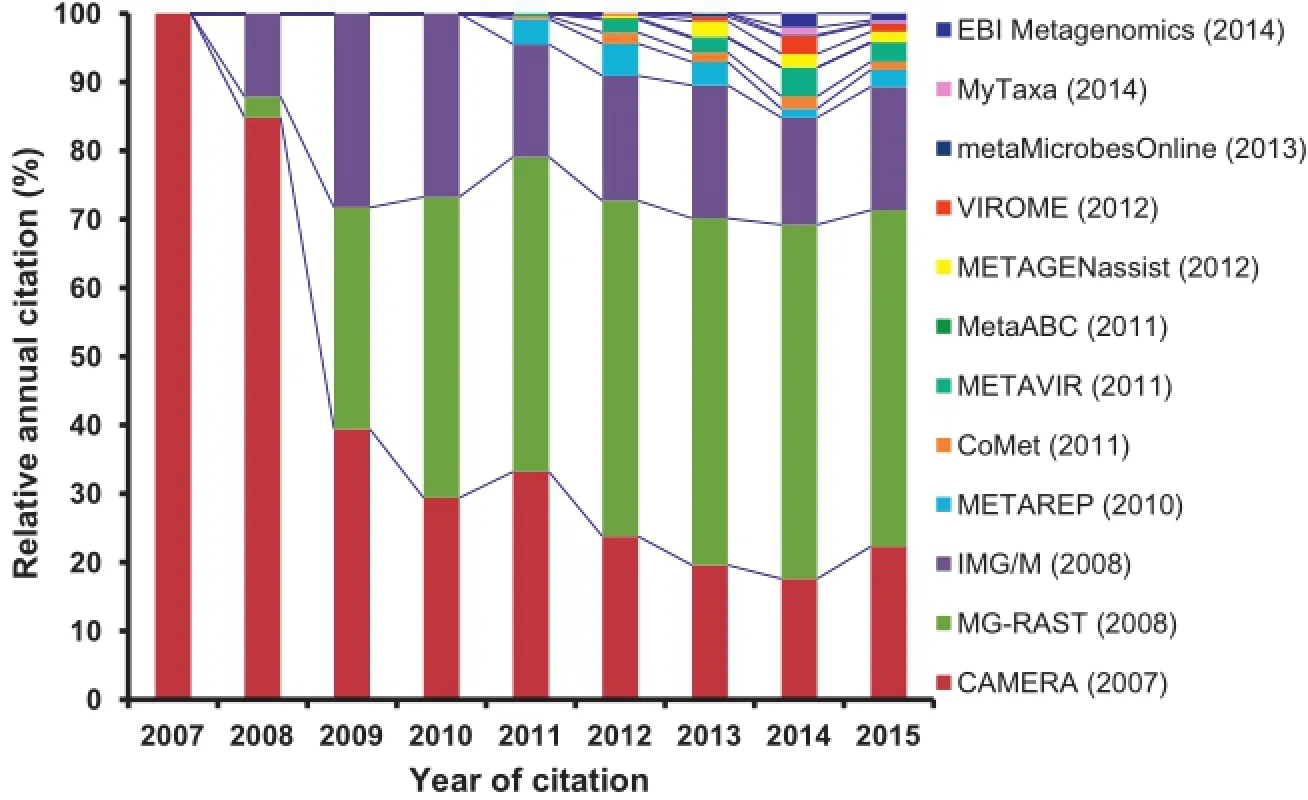
Figure 1 Relative citation of the metagenomics software tools from articles published in peer-reviewed scientific journals by October 2015 Year in the bracket in the legend box indicates the year of original release of the respective tool.Citation of each tool was tracked from Google Scholar.Totalcitation of alltools is considered as 100%in each year and relative percentage of citations of each toolper year was calculated in relative to the total citations of all tools in the respective year.

?
IMG/M
The software referred to as the Integrated Microbial Genomes and Metagenomes(IMG/M)is a data storage,management, and analysis system for metagenomes hosted by the Joint Genome Institute(JGI)of U.S.Department of Energy (DOE)[20].IMG/M integrates metagenome datasets with isolated microbialgenomes from the IMG system.It is a domainspecific tool of IMG compatible with sequencing data from microbialcommunities.IMG/Mallows for combined analyses with all available draft and complete genomes,plasmids,and viruses in the public domain[21,22].It provides free support for genomic and metagenomic data annotation,integration, and comparative analyses of integrated genomic and metagenomics data.The data content and analyticaltools are updated continually.As of January 2015,IMG/M contained a total of 5234 metagenome datasets with billions of genes,out of which 3193 metagenome datasets were publicly available.More than 13,945 registered users from 95 countries had gained access as of October 2015.Registered users can access IMG/Mdata content and analytical tools.Registered users can submit data online and maintain data’s privacy for two years before data move to the public domain.Furthermore,analysis and downloading ofmetagenomics datasets via JGI’s portals is also possible for registered users.Pre-processing,quality control, and annotation of input data are performed by JGI’s metagenome annotation system.The stored IMG/Mdata are annotated using multiple reference datasets to conduct analyses in three tiers:(i)phylogenetic composition,(ii)functionalor metabolic potentialwithin individualmicrobiomes,and(iii)comparisons across microbiomes.IMG/M provides support for such analyses by integrating metagenome datasets with isolated microbial genomes from the IMG system[23].IMG/M provides the outputdata in multiple-cluster forms,which facilitates its use as an online tool for environmental and organismal metagenomics analysis.In addition to providing integrated microbial metagenomes,IMG/M has developed the customized IMG/ M-HMP(Human Microbiome Project;http://www.hmpdacc. org),which provides analysis tools for the annotation of HMP-generated metagenome datasets in the reference of all publicly-available genomes and metagenomics samples in IMG[24].IMG/M attracts increasing number of researchers due to its in-depth descriptive analyses of metagenomes and genome extraction through binning of metagenomes.
METAREP
Metagenomics Reports(METAREP)is openly available software for comparative metagenomics analysis.It is a well-known open browser for annotation,analysis,and comparison of short reads or assemblies of metagenomics data [25].To upload input data,it requires only basic programming knowledge and the skill to operate the stand-alone version of METAREP.Users can simultaneously link a minimum of two datasets at various functional and taxonomic levels for comparative analyses through applying multiple clustering methods.For each of these clustering methods,METAREP provides a download option to export tab-delimited files for downstream analysis.The software integrates highly-scalable platforms Solr/Lucene,R,and CAKEPHP to compare huge volumes of metagenomic annotations.METAREP has been developed and maintained under utmost privacy by the J.C. Venter Institute(JCVI),and it is hosted at GitHub.Users are encouraged to suggest new features of interest for improving and updating the application.Recently,JCVI has updated the software with a better annotation process,clustering procedure,and input format.These improvements are practically demonstrated by the comparative analysis of shotgun metagenomics data from the HMP(http://www.hmpdacc.org/)[26].
CoMet
CoMet is a freely accessible tool for comparative metagenomics analysis.It is the right pipeline for rapid analysis of metagenomic short-read data[27].CoMet can easily handle metagenomics data generated by the 454 and Ion Torrent platforms.For the comparative analysis of metagenomic data,it accepts up to 20 metagenomic DNA sequencing files in multiple FASTA format as input;however zip files may also be submitted for faster uploading.Data sized up to 500 MB can be uploaded at a time for analysis through the easy-to-use submission interface.CoMet is specifically designed to compare the functional or metabolic profiles of multiple metagenomes in terms of Pfam protein domains and the associated Gene Ontology(GO)terms.CoMet combines ORF-finding and subsequent assignment of protein sequences to Pfam domain families with a comparative statistical analysis,providing a quick overview of putative functional differences in a set of metagenomic samples.Registration is not mandatory for job submission.The output comprises downloadable text files and also provides figures and a matrix of the statistical results.It has been the most used tool for comparative metagenomics study since 2011.
METAGENassist
METAGENassist is a freely available webserver for comparative metagenomic analysis.It can provide a bacterial census from diverse biomes and performs comprehensive multivariate statisticalanalyses on the data[28].The server accepts the CSV file format along with a wide variety of common formats generated by online and standalone metagenomic analysis tools such as mothur,MG-RAST,QIIME,and MEGAN.It is able to analyze marker-gene assisted as well as shotgun metagenomic data.METAGENassist is the only webserver to perform taxonomic-to-phenotypic mapping of the submitted data.Phenotypic information covering nearly 20 significant functional categories is automatically generated from the input data.The server uses univariate and multivariate dataprocessing and follows a multi-step workflow to cluster data into different layouts.Output is available in annotated colorful tables and graphs,which can be downloaded in PNG or PDF formats.
MetaABC
MetaABC is an integrated metagenomics platform for data adjustment,binning,and clustering.Its primary focus is data filtration and normalization techniques for improving taxonomic assignment in metagenomic analysis[29].MetaABC was developed with compatibility for the metagenomic data generated by both NGS and Sanger platforms.Currently, more than 50 metagenomic datasets have been stored in MetaABC.Users can simultaneously upload 2–20 SFF, FASTA,or FASTQ files for analysis in My MetaABC.After the uploading of data files,the user can select and execute all the steps for abundance profiling of microbes.The final outputs are delivered to a user-friendly interface and can be retrieved for downloading in different visualization formats. Although MetaABC has advantages,such as integrating a hierarchical clustering program for comparative analysis of metagenomes,it has limitations as it is not applicable for functional and metabolic profiling of metagenomes.Moreover,its data privacy is limited because unregistered use can also access the data,and sharing feature is not available.Furthermore,it does not accept large(>2 MB)input files.Instead,MetaABC provides a stand-alone version to deal with larger datasets.It has been freely available in the public domain for the last four years,though its citations are low and it does not seem to be gaining enough attention in the scientific field.
MyTaxa
MyTaxa is the latest novelplatform for the taxonomic assignment of metagenomic sequences with high accuracy.It has homology-based bioinformatics framework to classify metagenomic and genomic data with extraordinary precision[30]. MyTaxa employs all genes present in an unidentified sequence as classifiers,weighting each gene on the basis of its precomputed classifying power at a specified taxonomic level and frequency of horizontal gene transfer[30].MyTaxa is specifically applicable to consigning and classifying unknown genomic and metagenomic sequences.It consists of a standalone indexed database that is freely accessible and downloadable from MyTaxa’s website.The database contains parameters for the gene clusters that are employed in the online analysis for taxonomic classification.The database is regularly updated to achieve high sensitivity and specificity in taxonomic analyses by incorporating additional reference gene sequences that are available through isolated genome or sequencing projects.The toolis scalable to analyze large dataset as well as compatible with any new,faster algorithms besides homology search algorithms.In the web server,users can start MyTaxa analyses by supplying two files,a standard GFF file and a tabular output file.MyTaxa is equipped with a unique classification system based on the genome-aggregate average amino acid identity(AAI)concept.This classification system substantiates MyTaxa as an appropriate toolfor determining the degree of novelty of unknown metagenomic sequences.However,due to the absence of representative sequences for diverse species in the public domain,taxonomic assignment of the lowest taxa(i.e.,species)is a challenging task for MyTaxa.Moreover,its utility is hampered by the inability to resolve the functional or metabolic profiling of metagenomes.Finally,the BLASTx-translated metagenomic protein file is a prerequisite for operating the analytical tools, which turns out to be a key barrier for the new learners.
metaMicrobesOnline
The metaMicrobesOnline software is a free web portal for the phylogenetic study of genes from microbialgenomes and metagenomes.The metaMicrobesOnline analysis pipeline starts with contig assembly,gene calling,and translation into protein sequences,followed by scanning the sequence against recognized gene and protein domain families[31].Outputs of the scanning are used to add genes from the metagenomics data to a multiple sequence alignment for each gene family,followed by construction of phylogenetic trees for each gene/domain family using FastTree-2.Users can select the optimal approach for their data,from assembly and gene calling to gene-tree analysis.Genome-context browsing is available, combining metagenomic contigs and isolated genomes.Therefore,users can determine the contiguously-sequenced genome and search functionally-associated genes.metaMicrobesOnline contains a secured database of 155 metagenomes including 123 ecological and 32 host-associated metagenomes.It has been integrated with the MicrobesOnline(http://www.microbesonline.org)isolated genomes since July 2010,which holds thousands of genome sequences from all three domains of life [32].However,currently this tool is not being used widely and has not been frequently cited due to a lack of regular updates,the limited range of analyses,and restricted submission of new metagenomes.
EBI Metagenomics
EBI Metagenomics was the first resource developed in Europe by EMBL-EBI in 2011 for taxonomic,functional,and comparative metagenomics analysis[33].It contains QIIME-embedded tools for data management,analysis,storage,and sharing of metagenomes.Users must register to upload raw nucleotide reads that can remain private up to 2 years upon request,and reads are eventually stored with unique accession numbers in the European Nucleotide Archive(ENA).The uploaded datasets,along with essential details,comply with the Genomic Standards Consortium(GSC)and Minimum Information about any(X)Sequence(MIxS)for validation, so that the unambiguous data can be achieved and reused by the scientific community.Furthermore,users can submit raw sequencing reads generated from any NGS platforms through ENA’s Webin tool or ISAcreator.The EBI Metagenomics analysis pipeline includes trimming and quality checks to remove artifacts and streamline of the data.The processed reads are analyzed by rRNAselector and QIIME to predict taxa,followed by functional assignment by a composite protein database called InterProScan.EBI Metagenomics is well suited for the analysis of shotgun and marker-gene metagenomes.At present,it is mainly focused on shotgunmetagenomes;however,the use of rRNASelector for the extraction of rRNA from shotgun metagenomic datasets is a key feature for analyzing marker-gene metagenomes.The results of analyses are easily accessible via the EBI Metagenomics web interface and are downloadable in a variety of forms compatible with further analyses using online or standalone tools.Rapid comparative taxonomic and functional profiling of metadata is also made possible using EBI Metagenomics.The comparative analysis allows extraction and identification of common and unique information among various metagenomes.Due to the multiple advantages offered by EBI Metagenomics,it has quickly become a favorite tool among beginners.EBI Metagenomics is constantly updating the analysis pipeline with the development of additional analysis and visualization tools.Currently,it endeavors to establish a common collaborative platform for NGS computational setups.In near future,multi-omics data analysis should also be possible through the EBI Metagenomics platform.
CAMERA
The Community Cyberinfrastructure for Advanced Marine Microbial Ecology Research and Analysis(CAMERA)was developed in 2007 with the aim of monitoring microbial communities of the ocean and their response to environmental changes[34].In 2010,CAMERA expanded its mission to include all metagenomics data derived from different biomes by dropping the word‘‘marine”from its original name[35]. Currently,the Data Distribution Center(DDC)of CAMERA is no longer available at the original domain(http:// camera.calit2.net),but has been relocated to the iMicrobe Project(http://data.imicrobe.us),which holds 128 projects and 2660 samples as of October 2015.Like MG-RAST and IMG/M,CAMERA is a prominent large-scale database that processes,shares,and secures metagenomic datasets.The sharing of data and computed results is currently under the auspices of the GSC.CAMERA permitted the dataset’s publication and was the first web tool to support the GSC’s Minimal Information checklists for metadata.To submit data, users are required to register in the iPlant cyberinfrastructure through the Discovery Environment(DE)web interface. Registered users normally are allotted 100 GB of virtualspace, and an additional allocation of up to 1 TB can be granted upon request.The uploading and importing of huge datasets (>1.9 GB)within the DE is carried out either by Cyberduck for Mac and Windows users or by iDrop Desktop(iCommands)for LINUX users.Alternatively,small data files can be uploaded and imported within the DE using a simple URL.CAMERA’s fundamental task is to create a rich,distinctive data repository and bioinformatics tool resource that overcomes the unique challenges of metagenomics such as complexity,heterogeneity,and fragmented data.The database includes environmental metagenomic and genomic sequence data,associated environmental parameters,pre-computed search results,and software tools to support powerful crossanalysis of environmental samples.It holds a large number of samples from across the world;however,since 2011,its use and resulting citations have consistently decreased because ofthe technically lengthy and complicated data uploading process.The descriptive analysis of metagenomes is also not userfriendly.However:recently,it is incorporated into QIIME and offers rapid online cloud computing service with greater user involvement and tractability during the analysis process.
METAVIR
METAVIR is a freely available webserver exclusively designed to annotate the raw and assembled viral metagenomic sequences[36].Recently,a new framework,METAVIR 2, has been released that allows for comprehensive virome analysis[37].The server is useful for exploring viraldiversity from metagenomes and undertakes comparative analyses through pre-computed phylogenies of viral marker genes,rarefaction curves,and multivariate analyses constructed on k-mer signatures and BLAST-based comparisons.METAVIR 2 can handle assembled viromes made up of thousands of large contigs. As of October 2015,METAVIR holds 335 virome projects that comprise more than 64 million sequences from diverse samples.Registered users can upload their sequence datasets only in FASTA file in a private space.However,users can also submit large data with compressed files in zip,gzip,or tar.gz format.The output can be visualized in different interactive and dynamic forms,i.e.,tables,phylogenetic trees,recruitment plots,and maps.METAVIR has an easy-to-use submission interface and offers rapid in-depth virome comparison along with data privacy,which are key features that make it the most cited virome analysis pipeline.
VIROME
Viral Informatics Resource for Metagenome Exploration (VIROME)is a web-based application specifically designed for the analysisofmetagenomic data collected from viralassemblages occurring within different environmental contexts.It emphasizes the classification of metagenome viral sequences and putative ORFs based on homology search results against both known and environmental sequences[38].Submission of sequence data to VIROME is initiated through the web-application interface.After filling out a form,the user is contacted by Email correspondence with further instructions regarding sequence data transfer.It accepts multiple file formats,including FASTA,QUAL,FASTQ,and SFF. Metagenome libraries submitted for analysis at VIROME are blasted against the MetagenomesOnline database(MgOl),with a graphical interface and links to various protein metadata enabling exploration of the results.The VIROME workflow consists of two consecutive steps,sequence quality screening and sequence analysis against the UniVec database,followed by three parallel steps that include ORF-calling using MetaGeneAnnotator,known protein identification,and environmental protein characterization by subject databases such as MGOL and UniRef 100.Sequence quality trimming is run using a workflow managementsystem called Ergatis.Data from the sequence processing and BLAST analysis components are stored in a back-end MySQL database.An Adobe ColdFusion server handles communication between the MySQL database and the VIROME web application.VIROMEoffers downloadable outputthatcan be used as inputdata for multivariate analyses using third-party tools.The main hurdle to its widespread use is the length of time needed to analyze even a small-sized metagenome,usually severalweeks to months.
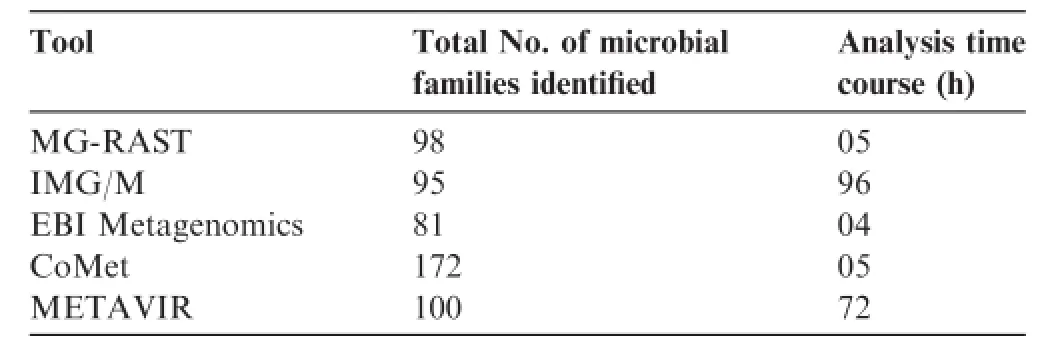
Table 2 Result of synthetic metagenome analysis using the five selected tools
Analysis of synthetic metagenome
Synthetic metagenome was analyzed using five selected tools to demonstrate the efficiency and analysis time course.Synthetic metagenome(Run accession:ERR340319)was downloaded from EMBL-EBI.It contains 32,782,167 bp incorporated in 90,385 raw sequences generated from 454 GS FLX Titanium sequencing.This synthetic metagenome was uploaded into MG-RAST,IMG/M,EBI Metagenomics,CoMetand METAVIR that were selected based on rating criteria for taxonomic assessment using online support provided by the software developers.The result showed the numbers of different microbial families reported from each tool are not exactly similar due to the different annotation pipelines used by these tools. Moreover,the analysis time required by the each toolwas also varied,because analysis depends on the number of running job in server and length of input sequences(Table 2).Though the remaining tools provided online help for analysis,it was not feasible to complete the data submission and taxonomic analysis of such a large synthetic metagenome within a short period of time by beginning users.
Concluding remarks
Structural and functional microbial profiling has rapidly become commonplace,thanks to the advent of NGS platforms.Additionally,the comparative metagenomics of various biomes quickly enlarged the field of microbial ecology.To decode the ecosystem comprehensively,new tools,frameworks,and hypotheses will be required for analysis,storage, visualization,and sharing of the dataset.A single platform is therefore not adequate for conducting holistic metagenomics analyses.Longer-read sequences,accurate assembly,and annotation pipelines are anticipated developments from impending metagenomics study.The present review has given an overview of the prevailing metagenomics analysis tools,and we expect more advanced software tools from their original developers in the near future.This review may serve as an atlas for rising researchers to choose the most appropriate webbased platforms for metagenomic data analysis.It is specifically intended to aid those organizations that have limited computational resources with which to analyze their data.
Competing interests
The authors have declared no competing interests.
Acknowledgments
The authors are thankful to BIT Virtual Centre,Patan Node, Hemchandracharya North Gujarat University,India for providing servers and computational facilities.
References
[1]Handelsman J,Rondon MR,Brady SF,Clardy J,Goodman RM. Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products.Chem Biol 1998;5: R245–9.
[2]Daniel R.The metagenomics of soil.Nat Rev Microbiol 2005;3:470–8.
[3]Thomas T,Gilbert J,Meyer F.Metagenomics–a guide from sampling to data analysis.Microb Inform Exp 2012;2:3.
[4]Dudhagara P,Ghelani A,Patel R,Chaudhari R,Bhatt S. Bacterial tag encoded FLX titanium amplicon pyrosequencing (bTEFAP)based assessment of prokaryotic diversity in metagenome of Lonar soda lake,India.Genom Data 2015;4:8–11.
[5]Ghelani A,Patel R,Mangrola A,Dudhagara P.Cultivationindependent comprehensive survey of bacterial diversity in Tulsi Shyam Hot Springs,India.Genom Data 2015;4:54–6.
[6]Dudhagara P,Ghelani A,Bhavsar S,Bhatt S.Metagenomic data of fungal internal transcribed Spacer and 18S rRNA gene sequences from Lonar lake sediment,India.Data Brief 2015;4:266–8.
[7]Carlos N,Tang YW,Pei Z.Pearls and pitfalls of genomics-based microbiome analysis.Emerg Microbes Infect 2012;1:e45.
[8]Oulas A,Pavloudi C,Polymenakou P,Pavlopoulos GA, Papanikolaou N,Kotoulas G,et al.Metagenomics:tools and insights for analyzing next-generation sequencing data derived from biodiversity studies.Bioinform Biol Insights 2015;5:75–88.
[9]Patel R,Mevada V,Prajapati D,Dudhagara P,Koringa P,Joshi CG.Metagenomic sequence of saline desert microbiota from wild ass sanctuary,Little Rann of Kutch,Gujarat,India.Genom Data 2015;3:137–9.
[10]Mangrola AV,Dudhagara P,Koringa P,Joshi CG,Patel RK. Shotgun metagenomic sequencing based microbial diversity assessment of Lasundra hot spring,India.Genom Data 2015;4:73–5.
[11]Singh AH,Doerks T,Letunic I,Raes J,Bork P.Discovering functionalnovelty in metagenomes:examples from light-mediated processes.J Bacteriol2009;191:32–41.
[12]Ounit R,Wanamaker S,Close TJ,Lonardi S.CLARK:fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers.BMC Genomics 2015;16:236.
[13]Mandal RS,Saha S,Das S.Metagenomic surveys of gut microbiota.Genomics Proteomics Bioinformatics 2015;13:148–58.
[14]Behnam E,Smith AD.The Amordad database engine for metagenomics.Bioinformatics 2014;30:2949–55.
[15]Overbeek R,Begley T,Butler RM,Choudhuri JV,Chuang HY, Cohoon M,etal.The subsystems approach to genome annotation and its use in the projectto annotate 1000 genomes.Nucleic Acids Res 2005;33:5691–702.
[16]Meyer F,Paarmann D,D’Souza M,Olson R,Glass EM,Kubal M,et al.The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes.BMC Bioinformatics 2008;9:386.
[17]Overbeek R,Olson R,Pusch GD,Olsen GJ,Davis JJ,Disz T, et al.The SEEDand the Rapid Annotation of microbialgenomes using Subsystems Technology(RAST).Nucleic Acids Res 2014;42:D206–14.
[18]Wilke A,Bischof J,Harrison T,Brettin T,D’Souza M,Gerlach W,et al.A RESTful API for accessing microbialcommunity data for MG-RAST.PLoS Comput Biol 2015;11:e1004008.
[19]Sun Q,Liu L,Wu L,Li W,Liu Q,Zhang J,et al.Web resources for microbial data.Genomics Proteomics Bioinformatics 2015;13:69–72.
[20]Markowitz VM,Ivanova NN,Szeto E,Palaniappan K,Chu K, Dalevi D,et al.IMG/M:a data management and analysis system for metagenomes.Nucleic Acids Res 2008;36:D534–8.
[21]Markowitz VM,Chen IM,Chu K,Szeto E,Palaniappan K, Grechkin Y,et al.IMG/M:the integrated metagenome data management and comparative analysis system.Nucleic Acids Res 2012;40:D123–9.
[22]Markowitz VM,Chen IM,Chu K,Szeto E,Palaniappan K,Pillay M,et al.IMG/M 4 version of the integrated metagenome comparative analysis system.Nucleic Acids Res 2014;42:D568–73.
[23]Markowitz VM,Chen IM,Palaniappan K,Chu K,Szeto E, Grechkin Y,et al.IMG:the Integrated Microbial Genomes database and comparative analysis system.Nucleic Acids Res 2012;40:D115–22.
[24]Markowitz VM,Chen IM,Chu K,Szeto E,Palaniappan K,Jacob B,et al.IMG/M-HMP:a metagenome comparative analysis system for the human microbiome project.PLoS One 2012;7: e40151.
[25]Goll J,Rusch DB,Tanenbaum DM,Thiagarajan M,LiK,Methe BA,et al.METAREP:JCVI metagenomics reports–an open source tool for high-performance comparative metagenomics. Bioinformatics 2010;26:2631–2.
[26]GollJ,Thiagarajan M,Abubucker S,Huttenhower C,Yooseph S, Methe′BA.A case study for large-scale human microbiome analysis using JCVI’s metagenomics reports(METAREP).PLoS One 2012;7:e29044.
[27]Lingner T,Asshauer KP,Schreiber F,Meinicke P.CoMet–a web server for comparative functional profiling of metagenomes. Nucleic Acids Res 2011;39:W518–23.
[28]Arndt D,Xia J,Liu Y,Zhou Y,Guo AC,Cruz JA,et al. METAGENassist:a comprehensive web server for comparative metagenomics.Nucleic Acids Res 2012;40:W88–95.
[29]Su CH,Hsu MT,Wang TY,Chiang S,Cheng JH,Weng FC,et al. MetaABC–an integrated metagenomics platform for data adjustment,binning and clustering.Bioinformatics 2011;27:2298–9.
[30]Luo C,Rodriguez-R LM,Konstantinidis KT.MyTaxa:an advanced taxonomic classifier for genomic and metagenomic sequences.Nucleic Acids Res 2014;42:e73.
[31]Chivian D,Dehal PS,Keller K,Arkin AP.MetaMicrobesOnline: phylogenomic analysis of microbial communities.Nucleic Acids Res 2013;41:D648–54.
[32]Dehal PS,Joachimiak MP,Price MN,Bates JT,Baumohl JK, Chivian D,et al.MicrobesOnline:an integrated portal for comparative and functional genomics.Nucleic Acids Res 2010;38:D396–400.
[33]Hunter S,Corbett M,Denise H,Fraser M,Gonzalez-Beltran A, Hunter C,et al.EBI metagenomics—a new resource for the analysis and archiving of metagenomic data.Nucleic Acids Res 2014;42:D600–6.
[34]SeshadriR,Kravitz SA,Smarr L,Gilna P,Frazier M.CAMERA: a community resource for metagenomics.PLoS Biol 2007;5:e75.
[35]Sun S,Chen J,Li W,Altintas I,Lin A,Peltier S,et al.Community cyberinfrastructure for Advanced Microbial Ecology Research and Analysis:the CAMERA resource.Nucleic Acids Res 2011;39: D546–51.
[36]Roux S,Faubladier M,Mahul A,Paulhe N,Bernard A,Debroas D,et al.METAVIR:a web server dedicated to virome analysis. Bioinformatics 2011;27:3074–5.
[37]Roux S,Tournayre J,Mahul A,Debroas D,Enault F. METAVIR 2:new tools for viral metagenome comparison and assembled virome analysis.BMC Bioinformatics 2014;15:76.
[38]Wommack KE,Bhavsar J,Polson SW,Chen J,Dumas M, Srinivasiah S,et al.VIROME:a standard operating procedure for analysis of viral metagenome sequences.Stand Genomic Sci 2012;6:421–33.
6 August 2015;revised 9 October 2015;accepted 13 October 2015
.
E-mail:dudhagarapr@gmail.com(Dudhagara P).aORCID:0000-0002-4041-6751.bORCID:0000-0002-7525-0624.cORCID:0000-0002-2318-5998.dORCID:0000-0003-4638-6925.eORCID:0000-0002-4364-2244.fORCID:0000-0002-4712-1921.
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
 Genomics,Proteomics & Bioinformatics2015年5期
Genomics,Proteomics & Bioinformatics2015年5期
- Genomics,Proteomics & Bioinformatics的其它文章
- CVTree3 Web Server for Whole-genome-based and Alignment-free Prokaryotic Phylogeny and Taxonomy
- Soil and Rhizosphere Associated Fungi in Gray Mangroves(Avicennia marina)from theRed Sea—A Metagenomic Approach
- First Insights into the Viral Communities of the Deep-sea Anoxic Brines of the Red Sea
- Marine Metagenome as A Resource for Novel Enzymes
- PacBio Sequencing and Its Applications
- The Value and Significance of Metagenomics of Marine Environments