
Cancer metabolic reprogramming: importance, main features,and potentials for precise targeted anti-cancer therapies
2014-09-26 06:00:58LiemMinhPhanSaiChingJimYeungMongHongLee
Cancer Biology & Medicine 2014年1期
Liem Minh Phan, Sai-Ching Jim Yeung, Mong-Hong Lee
1Department of Molecular and Cellular Oncology, 2Department of General Internal Medicine, Ambulatory Treatment and Emergency Care, 3Department of Endocrine Neoplasia and Hormonal Disorders, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, Texas, TX77030, USA
Cancer metabolism: major remodeling of cellular energy production and metabolic pathways in tumors
Cancer metabolic reprogramming has been recognized as one of the ten cancer hallmarks by Drs.Hanahan and Weinberg in their seminal review paper published in 20111.Some of the most striking changes of tumor cellular bioenergetics include elevation of glycolysis, increase in glutaminolytic flux, upregulation of amino acid and lipid metabolism, enhancement of mitochondrial biogenesis, induction of pentose phosphate pathway and macromolecule biosynthesis1-17.
Glycolysis
Compared to normal cells, cancer cells prefer using glycolysis even in normoxic condition18-20.This phenomenon is often referred as the Warburg effect because Dr.Otto Warburg discovered and reported these metabolic alterations in tumors in 1930 and 195618-20.Many decades later, numerous studies have provided additional insights into the abnormality of cancer metabolism.
In normal cells, glucose is catabolized to pyruvate, which can be later converted to acetyl-CoA to fuel the tricarboxylic acid cycle (TCA cycle, or Krebs cycle).TCA cycle generates NADH and FADH2to provide mitochondrial respiratory chain with electrons for energy production.This is an effective energy production mode since each glucose molecule can produce up to 36 ATP, largely thanks to mitochondrial respiration.In normal cells, glycolysis is prioritized only when oxygen supply is limited.In contrast, cancer cells preferentially use glycolysis even in the abundance of oxygen2,3,5,7,18-21.This is why tumor glycolysis is often called “aerobic glycolysis”, or the Warburg effect, to distinguish from the normal anaerobic glycolysis of healthy cells.
However, cancer cells have to compensate for the 18-fold lower efficacy of energy generation (glycolysis only makes 2 ATP per glucose molecule consumed while mitochondrial respiration can produce up to 36 ATP for each glucose molecule catabolized).Part of the solution is to upregulate glucose transporters, especially Glut1, Glut2, Glut3, and Glut4, to uptake more glucose5,22-24.In fact, the increase in glucose uptake is a major feature distinguishing tumor cells from normal cells.This difference has been widely exploited in Positron Emission Tomography (PET) imaging modality using radiolabeled analogs of glucose such as18F- fluorodeoxyglucose as a tracer to visualize tumors.
In addition, tumors remarkably elevate the expression of the majority of glycolytic enzymes.Major oncogenes such as Ras,Myc, and HIF-1α are reported to be master inducers of cancer glycolysis3,5,24.Many glycolytic enzymes are also upregulated in tumors because of elevated c-Myc and HIF-1α transcriptional activity and insufficient p53-mediated control.Indeed, c-Myc and HIF-1α are well recognized as two master inducers of glycolysis through direct or indirect transactivation of cancer glycolytic genes.These two transcription factors coordinate to promote the expression of key glycolytic enzymes such as HK2,PFK1, TPI1, LDHA, among others, in tumors2,3,5,7,21,25,26.In fact,most of glycolytic gene promoter areas contain consensus Myc and HIF-1α binding motifs.While HIF-1α is mainly functional in hypoxia, c-Myc is well known to promote its glycolytic target genes’ expression in normoxia.This coordination allows tumors to continuously drive glycolysis for supporting their rapid proliferation and accelerated biosynthesis2,3,7,11,15,16,21.
In contrast, p53 is known to suppress glucose uptake by directly inhibiting the transcription of glucose transporter Glut1 and Glut427,28and suppressing the expression of Glut328.Glut3 is an NF-κB target gene and p53 is found to block NF-κB activation, thereby considerably reducing Glut3 transcription and expression28.p53 also induces the expression of TIGAR to slow down cancer glycolytic flux29,30.Fructose 2,6-bisphosphate is an important allosteric activator of PFK1, a major glycolytic enzyme.Fructose 2,6-bisphosphate is produced by PFK2 from fructose 1-phosphate.By converting fructose 2,6-bisphosphate back to fructose 1-phosphate, TIGAR significantly slows down tumor glycolysis29,30.
The interaction among p53, c-Myc and HIF-1α has a decisive impact on the status of cancer glycolysis2,5,7,16,21,30.Many studies have characterized the communication between these three master regulators of cancer glycolysis and how the balance among these factors control the status of cancer metabolism.
On the other hand, the way tumor cells process pyruvate,the end product of glycolysis, is also different from normal cells.In normal cells, most of pyruvate is converted to acetyl-CoA to fuel the TCA cycle.Some pyruvate is used to produce alanine or lactate.In contrast, pyruvate-to-lactate is a preferred reaction in tumor cells due to the upregulation of lactate dehydrogenase A (LDHA).This reaction is bene ficial for cancer cells as it helps regenerate NADH to accelerate glycolysis2,3,5,11,25.Furthermore,lactate is secreted into tumor microenvironment via MCT4 transporter to fuel other cancer cells that do not have frequent access to nutrient supplies from blood stream.Lactate could be uptaken by MCT1 transporter and used by the TCA cycle for metabolism.The symbiosis of lactate-producing cancer cells and lactate-consuming tumor cells is an effective way for tumors’adaptation to the diverse and constantly changing conditions in tumors, which is caused by the leaky and poorly formed tumor blood vessel network7,31-33.Furthermore, converting pyruvate to lactate also reduces reactive oxygen species’ levels, thereby diminishing the intracellular oxidative stress in cancer cells and promoting tumors’ survival2,7.Moreover, lactate also lowers the pH of extracellular microenvironment and facilitates the activity of metalloproteases for breaking down extracellular matrix.Thus,lactate is an inducer of cancer invasion and metastasis34,35.
Importantly, glycolysis provides cancer cells with not only energy but also necessary precursors for biosynthesis, which is similar to stem cells’ metabolic profiles.Several glycolytic metabolites such as glucose-6-phosphate, dihydroxyacetone phosphate, among others, could be diverted into other metabolic pathways.For instance, glucose-6-phosphate is often consumed by pentose phosphate pathway to synthesize nucleotides and NADPH (a major reducing agent important for redox homeostasis and drug detoxifying reactions).Dihydroxyacetone phosphate could be used for lipid synthesis,which is important for assembling new organelles and cells to promote tumor growth and proliferation.Metabolites from glycolysis are also important materials for amino acid production and macromolecules synthesis, which is required for active cell division and large-scale biosynthetic programs2,3,5,7,16,36,37.In addition to their metabolic function, glycolytic enzymes play active roles in promoting cancer survival, metastasis, invasion,chromatin remodeling, gene expression regulation, and other essential cellular processes2,38.Thus targeting glycolytic enzymes’activities could be useful strategies for cancer therapy.
Glutaminolysis
In addition to glycolysis, many tumors also rely on glutaminolysis to fuel their cellular bioenergetics and metabolism.Glutaminolysis is a series of biochemical reactions catabolizing glutamine into downstream metabolites such as glutamate,α-ketoglutarate.The products of glutaminolysis are very important to fuel the TCA cycle of tumors.The intermediates of TCA cycles could be used for the synthesis of lipid, cholesterol,amino acids and other essential metabolites.Moreover, NADH and FADH2from the TCA cycle provide electrons for the electron transport chain of mitochondria to generate ATP.Thus,similar to glycolysis, glutaminolysis supplies cancer cells with not only ATP but also crucial precursors for continuous biosynthesis and accelerated proliferation3,5,13,15,16,22,25.
Glutaminolysis upregulation in tumors is mediated by c-Myc4,9,13,39.Multiple studies demonstrate that c-Myc promotes both glutamine uptake and the catabolic process of glutamine.In fact, c-Myc transactivates ASCT2 and SN2, two important glutamine transporters on cellular membrane9,40.c-Myc also suppresses miR-23a/b to upregulate GLS1 expression41,42.GLS1 is a major enzyme for glutaminolysis.Therefore, c-Myc is an important inducer of glutaminolysis in tumors.Interestingly,while promoting cancer metabolic reprogramming, c-Myc also renders cancer cells addicted to glutaminolysis, which opens a new therapeutic window to selectively suppress and eliminate cancer cells9,13-15,39,43.Therefore, targeting tumor glutaminolysis and c-Myc-induced-glutamine addiction is a promising anticancer metabolism therapy.
Pentose phosphate pathway
Pentose phosphate pathway (PPP) is a classical metabolic pathway consisting of two branches.In the oxidative arm, PPP converts glucose-6-phosphate, a glycolytic intermediate, into ribulose-5-phosphate and generates NADPH.NADPH is then used for glutathione production, detoxification reactions, and biosynthesis of lipids as well as other macromolecules.The nonoxidative PPP branch involves reversible carbon-exchanging reactions with the final products as fructose-6-phosphate and glyceraldehyde-3-phosphate.These metabolites can participate in glycolysis and downstream metabolic pathways44.PPP is commonly viewed as a line of defense counteracting reactive oxidative stress and producing ribose-5-phosphate for nucleotide synthesis.However, new studies suggest that PPP has important impacts on various aspects of cancer, including proliferation,apoptosis, invasion, drug resistance, and metastasis44.These exciting findings unveil PPP as a potential target for anti-cancer metabolism therapies.
Rapidlyproliferating cancer cells constantly demand nucleotides and other materials for biosynthesis.Therefore,by providing NAPDH and pentose phosphate for nucleotide synthesis, PPP is important and frequently upregulated in many types of tumors5,44.In fact, the activity of glucose-6-phosphate dehydrogenase (G6PD), a major PPP enzyme,increases in proliferating cancer cells45.G6PD, transketolase(TK) and other PPP enzymes are elevated in multiple types of cancer and facilitated tumors’ accelerated proliferation44,46,47.In addition, G6PD also promotes cancer survival by producing NADPH, a key tool for tumor cells to defend against oxidative stress, chemotherapy-induced cytotoxic damage, as well as for promoting biosynthesis44.Hence, G6PD function is tightly controlled by the tumor suppressor p53.Indeed, p53 associates with G6PD and prevents this enzyme from forming active dimer complexes48.It is noteworthy that G6PD is directly transactivated by HIF-1α49.The function of G6PD is strictly regulated in normal cells but highly activated in cancer cells,making G6PD a strong oncogene candidate44.Interestingly,G6PD and TK functions are both suppressed by resveratrol50,suggesting the usage of this natural product in cancer treatment and prevention.
While normal cells frequently rely on the oxidative branch of PPP for ribose-5-phosphate production; cancer cells use both arms, e.g., oxidative and non-oxidative, of PPP to generate ribose-5-phosphate for nucleic acid synthesis51-53.Furthermore, cancer cells can use ribose-5-phosphate in both de novo and salvage pathways to synthesize nucleotides.These flexible metabolic programs help cancer cells effectively adapt to constantly changing nutritional conditions of tumor microenvironment.
In addition, PPP also protects tumor cells from apoptosis by counteracting oxidative stress and facilitating DNA damage repair.In fact, nonsteroidal anti-inflammatory medications induce apoptosis and shrinkage of colon carcinoma and polyps by regulating PPP54.Moreover, G6PD inhibitors, e.g., DHEA and 6-AN, promote apoptosis in mouse fibroblasts and PC-12 neural cells while overexpression of G6PD protects cells from H2O2-induced cell death55.Knocking down of G6PD also increases oxidative stress-mediated toxicity in melanoma cells56.The vital role of PPP in protecting cells from programmed cell death is additionally proven in vivo such as in stem cells and peripheral blood mononuclear cells of patients lacking G6PD55,57,58.Interestingly, the cytoprotective function of PPP is not limited to defending against reactive oxygen species but also expands to helping DNA damage repair.Indeed, upon DNA damage, ATM quickly activates G6PD functions to accelerate PPP for quenching reactive oxygen species, increasing nucleotide synthesis and enabling effective DNA repair.Therefore, knocking down G6PD significantly impairs DNA damage repair ability59,60.Some other studies describe the impact of PPP on regulation of autophagy61, but the molecular mechanism is still not completely understood.
Surprisingly, PPP also induces tumor angiogenesis.Leopold et al.62and Pan et al.63reported the crosstalk between G6PD and VEGF and tight association between G6PD and angiogenesis.These studies show that VEGF stimulate G6PD expression via Src signaling and G6PD is important for VEGF-inducedendothelial cell migration by increasing the phosphorylation of VEGR receptor Flk-1/KDR.G6PD also increases the proangiogenic activity of endothelial NO by providing NADPH and stimulates Akt-induced activation of endothelial nitric oxide synthase (eNOS)62.
PPP additionally promotes tumor resistance to chemotherapy and radiation by multiple mechanisms.First, PPP provides cancer cells with NAPDH, a potent anti-oxidative agent that protects cancer cells from reactive oxygen species-induced cell death caused by chemotherapy and radiation44; Second,PPP facilitates DNA damage repair by providing material for nucleotide synthesis; Third, by shifting cancer metabolism away from mitochondrial respiration, PPP lowers the intracellular concentrations of reactive oxygen species, thereby increasing tumor endurance and survival during chemotherapy and radiation treatment; Fourth, NAPDH derived from PPP, is an important element for glutathione (GSH) generation.GSH is frequently used in detoxification reactions, enabling cancer resistance to a variety of chemotherapeutic agents.GSH conjugation to these xenobiotics also facilitates the activity of MDR1 and MDR2 to discard cytotoxic substances.Therefore,increase in G6PD expression and PPP flux increase intracellular GSH levels and reduce drug accumulation in cancer cells64.However, there are still many exceptions where PPP neither significantly contributes to drug resistance nor promotes the effect of certain chemotherapeutic agents in several cancer cell lines.This complexity requires more study to fully elucidate the contribution of PPP in protecting cells from anti-cancer treatments44.
In short, PPP is an important metabolic pathway providing cancer cells with NADPH, ribose-5-phosphate and other essential intermediates.NAPDH is crucial for counteracting oxidative stress and biosynthesis reactions.Ribose-5-phosphate is a major element for nucleotide synthesis.Interestingly, the impact of PPP on cancer cells is well beyond oxidative defense.Indeed, PPP upregulation promotes cancer cell survival,angiogenesis, proliferation, invasion, metastasis, and resistance to radiation and chemotherapies.Therefore, elevated and active PPP enzymes, for instance, TKTL or G6PD, are frequently observed in malignant, aggressive, proliferative and drugresistant cancer cells44.The new exciting discoveries about PPP open new therapeutic windows but also require more study to refine rational approaches for precise and effective targeting of this vital metabolic pathway in cancer cells.
Mitochondrial biogenesis
Another major change in cancer metabolism is the enhancement of mitochondrial biogenesis.In contrast to conventional concepts, mitochondria play very important roles in cancer because these vital organelles are the nexus of many essential metabolic pathways65.Mitochondria are not only the energy generators but also the factories synthesizing many indispensable molecules for cellular biosynthesis, growth and proliferation.Moreover, mitochondria additionally control the redox balance and Ca2+concentration, which is essential for cellular homeostasis65.Therefore, impairment of mitochondrial function or lack of mitochondrial biogenesis seriously suppresses tumorigenesis, tumor formation and growth65-71.Furthermore, in comparison with healthy and well differentiated cells, cancer cells frequently rewire their mitochondria to switch from a maximal energy production by mitochondrial electron transport chain to a well-adjusted balance among constant energy requirement,large-scale biogenesis programs and rapid cell proliferation65.Therefore, mitochondrial biogenesis and mitochondria are truly essential for tumor cells65.Hence, increase in mitochondria biogenesis is a significant advantage for cancer.
It is well established that c-Myc is a strong promoter of mitochondrial synthesis.In fact, c-Myc induces the expression of many nuclear-encoded mitochondrial genes.More importantly,c-Myc directly transactivates mitochondrial transcription factor A (TFAM).TFAM is a transcription factor that is indispensable for mitochondrial genes transcription and mitochondrial DNA replication72.In reality, TFAM promotes the right formation of mitochondrial transcription and replication complexes and facilitates the correct positioning of mitochondrial DNA for optimal gene transcription and proper mitochondrial DNA duplication65.As the synthesis of new mitochondrial components and replication of mitochondrial DNA are vital for de novo mitochondrial formation, c-Myc, indeed, plays a crucial role in elevating the number of mitochondria.As a consequence,lack of Myc expression and transactivational activity remarkably reduces mitochondrial mass as well as mitochondrial biogenesis,resulting in a severely suppressive impact on many metabolic pathways of cancer cells and tumorigenesis ultimately72.
Lipid synthesis
Increase in lipid metabolism is another remarkable feature of cancer metabolism.Lipids are important building blocks of new organelles and cells.Lipid synthesis is a multiple step process involving several enzymes such as ATP citrate lyase (ACLY),acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN),and stearoyl-CoA desaturase (SCD).This procedure starts with converting acetyl-CoA to malonyl-CoA by ACC.A series of condensation reactions by FASN results in saturated fatty acids.Fatty acids could be desaturated by SCD.Cancer cells frequently upregulate de novo fatty acid synthesis to satisfy their demands for lipids73-75.FASN elevation is observed in breast, prostate and other types of cancer73,76-79.FASN is a target gene of HIF-1α and frequently overexpressed in an Akt and SREBP1-dependent manner80.ACLY, often activated by Akt81, is indispensable for tumor transformation and formation both in vitro and in vivo81,82.ACC is also very important for tumorigenesis as inhibition of ACC stops cancer growth and induces apoptosis of prostate cancer cells83.Furthermore, cancer cells often have higher lipid accumulation in form of lipid droplets in relative to normal cells84.
Cholesterol synthesis, or the mevalonate pathway, is also an important aspect of lipid biosynthesis because cholesterol is a major component of membranes controlling the membrane fluidity and formation of lipid rafts.Cholesterol is vital for activation of Ras-Raf signaling pathway85and deregulation of cholesterol synthesis is correlated with tumorigenic transformation86.Interestingly, statin-mediated inhibition of HMGCR, an important enzyme of the mevalonate pathway,considerably ameliorates the effectiveness of chemotherapies in acute myeloid leukemia87, hepatocellular carcinoma88, and other types of cancer through epigenetic pattern modi fication89.
The sterol regulatory element-binding proteins (SREBPs)are the main transcription factors controlling the expression of most of enzymes involved in fatty acid and cholesterol synthesis.SREBPs are helix-loop-helix 125 kDa proteins that require a protein cleavage at the endoplasmic reticulum for activation73.While SREBP1 controls fatty acid, triacylglycerol and phospholipid synthesis, SREBP2 regulates cholesterol generation90.SREBPs are controlled by tumor suppressors and oncogenes.AMPK, for instance, inhibits SREBP activation91and suppresses ACC91, thereby keeping lipid synthesis in check.Loss of pRb upregulates SREBP1 and SREBP2, thereby activating Ras signaling92.p53 mutants, on the other hand, coordinates with SREBP to transactivate cholesterol-synthesizing enzymes93.Of note, SREBP1 and SREBP2 are often overexpressed in cancer76and play an important role in cancer cell survival94.
At the organism level, excessive lipid synthesis contributes to tumorigenesis.It has been well documented that obesity increases the risk of cancer73.In fact, excessive lipid concentrations in liver and muscle cells induce insulin resistance by impairing insulin signaling and reducing glucose uptake.Insulin resistance forces pancreatic cells to secrete more insulin and insulin-like growth factors, which is very bene ficial for cancer proliferation and survival95-97.Obesity also increases inflammation, which contributes to insulin resistance and tumorigenesis98.Dietary restriction may reverse these tumorigenic trends but in certain scenarios, especially when PI3K/Akt signaling is overactivated, the tumor-suppressing impact of dietary limitation decreased99.A possible explanation is that nutrient restriction may reduce the levels of circulating insulin and insulin-like growth factors.However, the constitutive activation of PI3K/Akt may compensate for that insulin signaling decrease100.
Fatty acid oxidation
While glycolysis, glutaminolysis, fatty acid synthesis have been well characterized during the past few decades; fatty acid oxidation (FAO) still remains a little known metabolic pathway.However, recent studies have demonstrated the important contribution of FAO to tumorigenesis101.
Fatty acids are a rich energy source that can yield to up to two times more ATP than carbohydrates when needed.Fatty acids could be oxidized in mitochondria or by cytoplasmic lipophagy,a new fatty acid catabolic process102.FAO is a repeated multiround process leading to the production of acetyl CoA, NADH,and FADH2in each cycle.Acetyl-CoA can be imported into TCA cycle to generate more NADH and FADH2, which subsequently fuel mitochondrial respiration chain for ATP production.Acetyl-CoA can also fuel TCA cycle for synthesis of citrate.Citratederived isocitrate and malate can be respectively converted to α-ketoglutarate by IDH1 or pyruvate by malic enzyme (ME1)102.Both reactions generate NADPH, which plays a very important role in maintaining redox homeostasis, inducing cell survival,enabling xenobiotics detoxi fication and promoting biosynthesis for cell growth and division103.Of note, NAPDH is crucial for the function of many anabolic enzymes to sustain large-scale biosynthetic programs in many cancer cells.
NAPDH derived from FAO is very important for cancer cells to quench reactive oxidative stress.For instance, blocking glioma tumor’s FAO leads to rapid depletion of NADPH, surge of reactive oxidative species’ concentrations and increase in apoptosis104.NADPH produced by FAO is also relevant to the maintenance of hematopoietic stem cells because these cells are very sensitive and vulnerable to reactive oxidative stress.In fact,increased reactive oxygen species levels inhibit hematopoietic stem cells’ self-renewal and leads to cell differentiation105-107.Jeon et al.108reported that LKB1-APMK regulates the balance between NADPH consumption by fatty acid synthesis and NAPDH production by FAO.In fact, AMPK blocks fatty acid synthesis in tumors by phosphorylating and inactivating acetyl-CoA carboxylase (ACC)109, antagonizing PPAR signal transduction110and regulating CTP1C expression111.Therefore,AMPK is a potent inhibitor of fatty acid synthesis in cancer cells.
Needless for further emphasis, ATP is by large one of the most important molecules for cancer cells.Due to its rapid proliferation and accelerated activities, tumors are almost constantly in high demand for ATP.ATP is the most frequently used energy currency and a major material for phosphorylation reactions, an essential mode of cellular signal transduction and protein modi fication.ATP is also an indispensable element for DNA and RNA replication and repair.The function of MDR1 and other ABC pumps on cellular membrane, a major tumors’line of defense against chemotherapy, absolutely requires ATP.
Recently, ATP production by FAO has been shown to prevent anoikis, a type of cell death due to loss of attachment to extracellular matrix although the molecular mechanism still remains unclear and warrants more study103,112.The Pandolfigroup113also reported that the promyelocytic leukemia (PML)protein induced FAO by activating peroxisome-proliferatoractivated receptors (PPARs), leading to poor survival and clinical outcomes of breast cancer patients.Moreover, Tak Mak’s lab111additionally found that carnitine palmitoyl-transferase 1 isoform C (CPT1C) is an oncogene that induces cancer growth,ATP production, FAO and confers resistance to mTORC1 inhibitors.CPT1 proteins mediate the import of fatty acids into mitochondria for FAO reactions.CPT1 links carnitine to fatty acids and transports the conjugated products (acyl-carnitines)into mitochondria.Therefore, the oncogenic property of CPT1C is a good example illustrating the potential of FAO in tumorigenesis.
FAO is also important in ensuring cancer cell survival in a manner that is independent of ATP production101.In fact, CPT1 proteins suppress the pro-apoptotic function of Bax and Bak by modulating the formation of mitochondrial permeability transition pores and reducing cytochrome c release114,115.The results from Samudio et al.116and Vickers group117additionally indicate that FAO can promote cancer cell survival by preventing a cytotoxic intracellular surge of fatty acid concentrations.On the other hand, several groups show that the increase in reactive oxygen species due to FAO-induced mitochondrial respiration could be harmful for leukemia cells.However, this toxicity could be resolved by upregulating uncoupling protein 2 and 3 (UCP2, UCP3) that effectively dissipate the gradient proton in mitochondria and decrease mitochondrial oxidative phosphorylation efficiency118.
Thus, fatty acid oxidation promotes cancer cell survival, and provides tumors with necessary energy and precursors.The new findings about FAO reveal fascinating understandings about cancer metabolic reprogramming and unveil very promising opportunities for anti-cancer therapeutic approaches.However,additional knowledge is needed to successfully develop effective therapies targeting this important catabolic process in cancer.
Interestingly, Hu et al.119has recently completed a massive meta-analysis of over 2,500 microarrays including 22 types of cancer to compare the metabolic gene expression landscape of tumors relative to that of corresponding normal tissues.From this comprehensive transcriptomics analysis, three important observations have been reported: (1) despite the process of tumor evolution, there is still a significant degree of similarity in the gene expression metabolic profiles of tumors in comparison with those of the normal tissues where tumors originate; (2) the metabolic gene expression landscape across different types of tumors is heterogeneous.However, glycolysis,nucleotide synthesis, aminoacyl-tRNA synthesis, and pentose phosphate pathway are consistently upregulated and increasingly important in actively proliferating cancer cells; (3) hundreds of metabolic isoenzymes demonstrate remarkable and cancerspecific expression alterations, representing new significant therapeutic opportunities for anti-cancer metabolism therapies.These isoenzymes are important for cancer.Some enzymes such as isocitrate dehydrogenase and fumarate dehydratase,may even imitate or aggravate the impact of tumorigenic genetic mutations119.
In short, metabolic reprogramming is an important cancer hallmark characterized by the upregulation of glycolysis,glutaminolysis, lipid metabolism, mitochondrial biogenesis,pentose phosphate pathway as well as other biosynthetic and bioenergetic pathways.These cancer metabolic programs provide tumor cells with not only necessary energy but also crucial materials to support large-scale biosynthesis, rapid proliferation,survival, invasion, metastasis and resistance to anti-cancer therapies.Therefore, exploiting the unique features of cancer metabolism for cancer detection, treatment and monitoring is a very promising trend in cancer therapeutics, diagnosis and prevention.
Cancer metabolism and diagnostic imaging
The distinguished features of cancer metabolism have been extensively exploited for initial diagnosis, staging disease,monitoring tumor responses to therapies, and detecting cancer recurrence120.Therefore, nowadays, metabolic molecular imaging plays an indispensable role in clinical oncology.These diagnostic methods are non-invasive and can accurately detect the changes in selective biologic processes of tumors compared to normal surrounding tissues both at the initial tumor sites and metastatic locations over an extended period of time.The information provided by advanced imaging modalities such as PET, magnetic resonance spectroscopy imaging (MRSI), magnetic resonance imaging (MRI), is very valuable for cancer detection, prevention,and treatment120.
Positron emission tomography
PET is frequently combined with X-ray computed tomography(CT) to provide detailed information about cancer and anatomic locations of tumors.PET measures the signals of radiolabeled tracers taken up by cancer cells.PET is safe and widely used in clinics because the small amount of imaging probes doesn’t interfere with normal physiological processes.18F-fluoro-2-deoxyglucose (FDG) is the most commonly used PET imaging material.Since most of tumors have a high glycolytic flux,elevated glucose uptake and increased hexokinase function, they will often have higher FDG signals relative to normal tissues.After being imported into tumor cells, FDG is phosphorylated by hexokinase but phosphorylated FDG cannot be further catabolized by glycolytic pathway.Therefore, phosphorylated FDG molecules are accumulated in tumors and can be detected by PET scanners.In clinics, FDG-PET scan is commonly used for determining cancer stages, identifying cancer recurrence and assessing tumor response to anti-cancer therapies121,122.
In addition to upregulated glycolysis, other patterns of cancer metabolism are also used for molecular oncology imaging using PET scan.Choline, for example, is frequently absorbed by tumor cells and used for new cellular membrane biosynthesis,an important process for cell division.Therefore11C and18F radiolabeled choline tracers have been successfully applied in hepatocellular carcinoma, lung, brain, and prostate cancer diagnosis123-126.Similarly, 3'-deoxy-3'-18F-fluorothymidine is often used to monitor cancer cell proliferation in vivo.3'-deoxy-3'-18F-fluorothymidine is a thymidine analog and frequently phosphorylated by thymidine kinase 1.This enzyme is highly active in rapidly dividing cells, e.g., tumor cells, especially in S phase.Thus, 3'-deoxy-3'-18F-fluorothymidine PET can identify and measure tumor malignancy, tracking the efficacy of anticancer therapies127.Many other tracers are also used in PET imaging modality to monitor specific biological processes of tumors.For instance,68Ga-DOTATOC, a high-affinity ligand for somatostatin receptor 2, is used to detect neuroendocrine cancer masses128.16-α-18F- fluoro-17β-estradiol is used to quantify ERα and ERβ expression129.Tumor angiogenesis and the effectiveness of anti-angiogenic therapeutic agents are measured by tracers containing arginine-glycine-aspartic acid-peptide ligands.These ligands associate with αvβ3integrin whose expression is elevated on newly formed blood vessels130.Nitroimidazole is also exploited to image hypoxic areas where tumors are frequently located131.
In summary, PET with radiolabeled metabolic tracers is certainly a valuable and powerful imaging method with vast applications in clinical oncology.This diagnostic modality is continuously improved and more advanced tracers are in development.However, radiation is still a major concern for PET and its tracers.The radiation containment and safety are also other significant issues for PET application in clinics120.In addition, a complete understanding about cancer metabolic patterns and bioenergetics programs is crucial to continuously innovate metabolic tracers-based PET scan imaging.
The combination of MRI and MRSI
MRI and MRSI are often combined in clinical oncology diagnostics because1H MRSI is easily compatible with currently available MRI scanners in clinics132-134.1H MRSI has a high sensitivity and could be applied on a number of tracers120.During the past few years, MRSI has made significant advances and rapidly become a reliable imaging modality.A number of1H tracers have been successfully developed.For instance,1H choline-containing metabolites are employed to measure tumor malignancy.Choline is an important component of cellular membrane.Higher choline concentrations are detected in aggressive and malignant tumors in comparison with benign and normal tissues135,136.In fact, many breast tumors contain a large amount of choline while benign tumor masses often have low levels of choline135,136.Since the accumulation of choline is associated with increased cell proliferation in brain, breast,cervical and prostate cancers133,137-139, choline availability could be used as a marker for predicting tumor histologic grade,aggressiveness, and even response to anti-cancer therapies with low unspecific detection rates120,139.Moreover, as brain tumors often have increased choline concentrations and diminished levels of N-acetyl aspartate, the ratio of choline/N-acetyl aspartate has been used to evaluate the aggressiveness of several types of brain tumors140-142.Choline/creatinine ratio measurement is also a valuable indicator of oligodendroglial cancer grade143.
13C tracers are emerging important diagnostic probes although their application is still at early stages.Recently, Nelson et al.144reported a successful preclinical study and phase I clinical trial results with 31 prostate cancer patients.This is a pioneer project examining the applicability and safety of hyperpolarized13C pyruvate tracers to monitor and evaluate the metabolic changes,especially13C pyruvate-to-13C lactate flux, of prostate tumors in patients.This technique enabled a 10,000-fold increase in signals compared to regular MRI.Results were very promising with excellent safety pro files and accurate detection of13C pyruvateto-13C lactate flux in tumor areas that were subsequently proven by biopsy-based pathological and histological analyses.The success of this pioneer study paves a new way for non-invasive,safe, precise, and sensitive cancer diagnosis as well as tumor monitoring.A number of new types of13C metabolic tracers are under development and will certainly play a major role in cancer detection and imaging in future.
Poor spatial resolution used to be a challenge for MRSI133,138,145,146, but new advances and ongoing technological improvements are addressing this limiting factor, making MRSI a promising adjunct to MRI.Combining conventional MRI with MRSI will enable accurate, safe and non-invasive characterization of tumors.This new diagnostic strategy is especially important when collecting lesion biopsies is risky, painful and difficult.Thus, in future, this new combinatory imaging modality will reduce patients’ discomfort, concern, risk, pain, and avoid unnecessary invasive diagnostic procedures while increasing the accuracy, reliability and sensitivity of diagnosis120.
In summary, diagnostic imaging plays a crucial role in cancer detection and treatment.Exploiting the unique features of cancer metabolism is a very promising direction for developing novel diagnosis methods to accurately detect cancer lesions even at early stages and precisely monitor tumors’ responses to therapies.
Therapeutic implications
Given the vital role of metabolic reprogramming for tumorigenesis, targeting cancer bioenergetics is a very promising and rapidly rising direction for anti-cancer therapy development nowadays.Many compounds have been developed to selectively and effectively inhibit metabolic enzymes that are important for tumors.These inhibitors are currently at various stages of clinical trial process and we expect to see them in clinics within five to ten years from now.
One of the most common trends in anti-cancer metabolism therapies is to inhibit enzymes that are exclusively or mostly expressed or used in tumor cells.This therapeutic strategy would effectively eliminate tumors while minimizing damage to normal cells.Several groups have successfully developed inhibitors for Glutaminase 1 (GLS1), a glutaminase isoform that is highly upregulated in cancer cells, and proved the efficacy of blocking GLS1 in cancer treatment147,148.This tactic bases on previous studies showing a significant dependence of c-Myc-overexpressing cancer cells on glutaminolysis9,11-15,25,149.Similarly, modulating the activity of PKM2, a glycolytic enzyme that is frequently elevated in tumors, is also a promising therapy150,151.Fatty synthase (FASN)is important for palmitate synthesis and this enzyme’s expression is elevated in many tumors.Therefore, several groups have developed FASN inhibitors to target tumorigenesis75,152.Many inhibitors for HIF and HIF targets, for instance monocarboxylate transporter MCT4 and carbonic anhydrase IX (CAIX) are also potential anti-cancer drugs in future153-156.Similarly, MCT1 and carbonic anhydrase XII are targets of great potential153,154.MCT1 and MCT4 suppressors inhibit cancer growth in vitro and in vivo and invasion in vitro157-160.In fact, interfering with lactate transport by MCT1 and MCT4 inhibitors has been shown to induce tumor cell starvation and subsequent apoptosis158.
Blocking lactate production using dichloroacetate (DCA)shows promising results with minor side effects in early phase clinical trials, especially in glioblastoma patients161,162.DCA is found to promote pyruvate-to-acetyl-CoA flux and reduce pyruvate-to-lactate conversion, thereby inducing tumor shrinkage and apoptosis in vivo161-163.Clinical trial data show that DCA also suppresses tumor angiogenesis, blocks HIF1-α signaling and activates p53 in glioblastoma multiforme patients161.Initial studies additionally find that DCA inhibits pyruvate dehydrogenase kinase 1 (PDK1) activity and thereby activating the function of pyruvate dehydrogenase 1 (PDH1),an important enzyme catalyzing the pyruvate-to-acetyl-CoA biochemical reaction162,163.However, more and larger clinical studies are needed to fully elucidate the mechanism of action of this interesting compound and further evaluate its efficacy in cancer patients.
Glycolysis inhibitors are also of interest for many groups and pharmaceutical companies.For instance, FX11, a selective suppressor of lactate dehydrogenase A (LDHA) activity, was tested by Le et al.164and is currently studied by National Cancer Institute’s Experimental Therapeutics Program (NExT).2-deoxyglucose (2-DG) is among the most advanced cancer metabolism inhibitors in clinical trials (Phase II).2-DG reversibly inhibits hexokinase to block glycolysis.2-DG usage in combination with radiation demonstrates a good safety profile and slightly improves survival of glioblastoma multiforme patients165,166.However, the effects of 2-DG may be limited by high concentration of glucose because 2-DG-mediated inhibition of hexokinase is reversible.
Inhibiting mutant isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) is a remarkable therapeutic approach because these mutant enzymes have distinct activities compared to normal IDH1 and IDH2 in the healthy cells.On the other hand, metformin, a common anti-diabetics medication,has demonstrated very promising impact in cancer treatment.It is well known that metformin inhibits mitochondrial complex I of liver cells, thereby decreasing ATP production.Lack of ATP subsequently stimulates LKB1-AMPK pathway and blocks gluconeogenesis, leading to lower blood glucose concentrations, improved sensitivity to insulin and diminished insulin production167.It is currently unclear whether metformin improves cancer patient clinical outcomes by lowering blood glucose levels and insulin/insulin-like growth factors generation or by directly targeting cancer cells.Nevertheless, the usage of metformin has been well documented to ameliorate cancer patient survival168,169and metformin are harmful for cancer stem cells170.Clinical trials testing the impact of metformin on cancer in patients are ongoing (Table 1).
Importantly, there is also an urgent need to develop effective inhibitors to target the key inducers of cancer metabolic reprogramming such as c-Myc and Ras.Ras mutations and c-Myc upregulation are frequent in many common types of cancer and these dysregulations are major drivers of tumorigenesis and resistance to therapies171,172.However, despite our relentless efforts, effectively and directly inhibiting Ras and c-Myc still requires a lot more study because these two proteins are currently undruggable targets.Interestingly, several preclinical research projects show that targeting metabolic enzymes significantly inhibits tumors carrying Ras mutation and c-Myc overexpression9,173.In fact, suppressing glycolysis andglutaminolysis remarkably antagonizes the growth of tumors bearing those genetic alterations9,164,174,175.These observations imply a new way to treat tumors carrying genetic mutations that can’t be directly targeted.
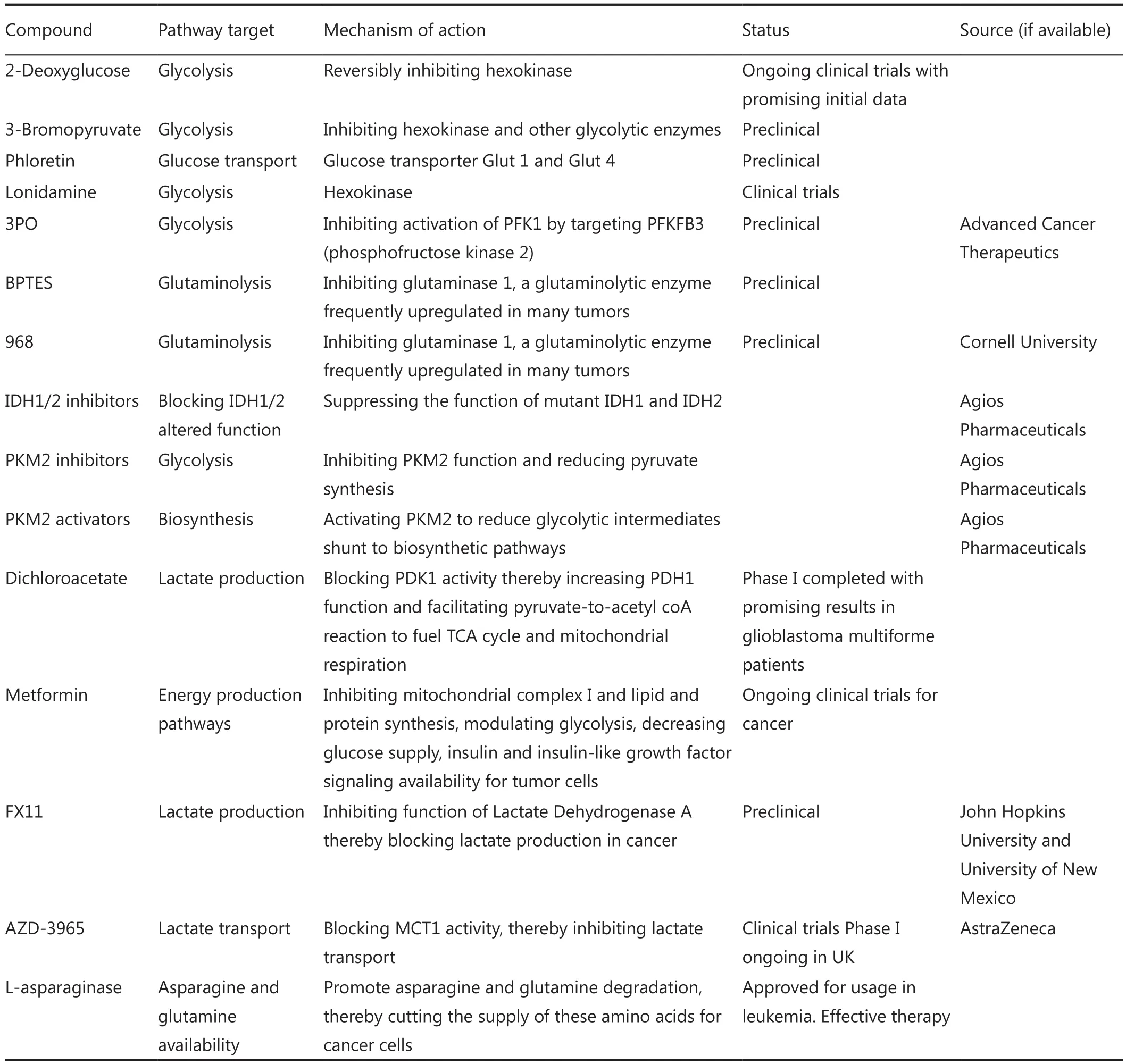
Table 1 List of several potential anti-cancer metabolism compounds
Another striking example of successful anti-cancer metabolism therapies is L-asparaginase.L-asparaginase mediates deamination reactions to degrade asparagine into aspartic acid176, thereby reducing asparagine availability to cancer cells and suppressing their growth177,178.This therapy is very effective for acute lymphoblastic leukemia (ALL) and related leukemia subtypes because ALL cells are unable to synthesize asparagine179.Therefore, these cancer cells have to rely on extracellular asparagine sources and become very vulnerable when asparagine supplies are limited.
However, lymphocytes, especially T cells, have similar metabolic programs as those in tumor cells.For instance,lymphocytes also depends on glutamine metabolism180,suggesting that systematically targeting glutaminolysis for cancer treatment may severely affect adaptive immune responses and also innate immunity to a certain degree.These metabolic similarities between cancer cells and lymphocytes explain why many agents targeting cancer metabolism are also strong immunosuppressants.For instance, cyclosporine, a potent anti-cancer drug that inhibits mTOR, significantly suppresses immune system.Suppressor of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme responsible for nicotinamide adenine dinucleotide (NAD+)regeneration, is poisonous to lymphocytes181.In fact, early clinical trials data show that FK866, a NAD+synthesis inhibitor, leads to mild lymphopenia and severe thrombocytopenia182.
These findings suggest that immunosuppression could be a challenge for therapies designed to target cancer cells’bioenergetics as the Achilles’ heel of tumors.Nevertheless, there is still a significant therapeutic window for anti-cancer metabolic therapies.We just need to identify the key differences in the bioenergetics patterns of tumors and those of healthy cells in order to optimize our therapies for precisely inhibiting the unique metabolic targets in cancer cells.A significant example is to use BPTES to selectively block GLS1, a glutaminase enzyme isoform that is crucial for cancer cells and specifically upregulated in tumors147,148.
Conclusion
Metabolic reprogramming is a major hallmark of cancer, which is characterized by upregulated glycolysis, glutaminolysis,lipid metabolism, pentose phosphate pathway, mitochondrial biogenesis, among others.These metabolic programs provide cancer cells with not only energy but also vital metabolites to support large-scale biosynthesis, continuous proliferation and other major processes of tumorigenesis.Potent oncogenes as c-Myc, HIF1α, Ras and PI3K/Akt are important promoters of cancer metabolic alterations.In contrast, major tumor suppressors such as p53 and LKB1/AMPK antagonize those changes and keep cellular metabolism in check (Figure 1 and Figure 2).Rfewiring metabolism is very beneficial for tumor survival, invasion,metastasis, growth, angiogenesis, proliferation and resistance to anti-cancer therapies.Although there is still much to study and discover, recent remarkable advances in this field have unveiled exciting therapeutic windows to precisely and effectively target cancer metabolism and bioenergetics (Figure 3).It is expected that anti-cancer metabolism therapies will play an important role in clinical oncology within five or ten years.
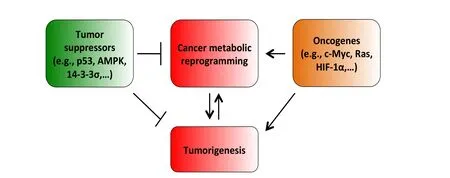
Figure 1 The impacts of tumor suppressors and oncogenes on cancer metabolic reprogramming, an important cancer hallmark.Cancer metabolic alterations are the results of oncogene activation and mutant metabolic enzymes.Cancer metabolic reprogramming promotes tumorigenesis by facilitating and enabling rapid proliferation, survival, invasion, metastasis, resistance to therapies and other central cellular processes of tumorigenesis.On the other hand, as tumorigenesis advances, cancer cells acquire more mutations and changes that further enhance metabolic reprogramming and, in turn, accelerate tumor growth, proliferation and progression.Tumor suppressors, for instance, p53, and AMPK, exert their suppressive regulation on cancer metabolic alterations by blocking the function, activation and expression of essential cancer metabolic genes.Our recent results also show that 14-3-3σ, a downstream target gene of p53, effectively opposes and reverses cancer metabolic reprogramming.Our data indicate that 14-3-3σ accelerates the degradation of c-Myc, an important transcription factor promoting cancer metabolic reprogramming183.In contrast, oncogenes such as c-Myc, HIF-1α, Ras, and Akt are major inducers of tumor bioenergetics alterations by upregulating the expression or activation of key metabolic enzymes such as HK2, GLS1, LDHA, among others.The balance between tumor suppressors and oncogenes has a decisive impact on the status of cancer metabolism.
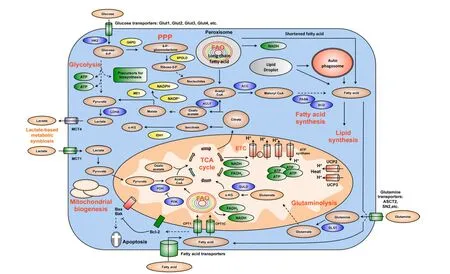
Figure 2 Summary of key changes in cancer metabolic reprogramming.Cancer metabolic reprogramming is characterized by enhanced glycolysis, PPP, lipid metabolism, glutaminolysis, mitochondrial biogenesis, among others.These pathways provide cancer cells with not only essential energy but also important precursors to support large-scale biosynthesis, rapid proliferation, continuous growth, tissue invasion, metastasis, survival and resistance to anti-cancer therapies.For instance, glycolysis generates 2 ATP per glucose consumed and provides materials for PPP and other biosynthetic programs.Similarly, PPP supplies tumors with ribose-5-phosphate and NADPH.Ribose-5-phosphate is a major element for nucleotide synthesis, which is used in DNA replication, RNA synthesis,and DNA damage repair, among others.NADPH is a key line of defense counteracting oxidative stress and a crucial metabolite for a number of biosynthesis reactions.NADPH is produced by 4 biochemical reactions mediated by G6PD, 6PLGD, ME1 and IDH1.In addition, fatty acid synthesis is indispensable for formation of new cellular membranes and proliferation.A number of fatty acid synthesis enzymes such as ACC, ACLY and FASN are upregulated or activated by oncogenes such as c-Myc, HIF-1α, Akt, among others.On the other hand, FAO is also important for cancer cells because it generates energy, NADPH and other necessary metabolites.Fatty acids are imported into mitochondria by CPT1 and oxidized to generate acetyl-CoA.Acetyl-CoA fuels the TCA cycle to generate NADH and FADH2.The latter metabolites donate electrons to mitochondrial ETC for ATP generation.CPT1 also antagonizes Bax and Bad-mediated apoptosis by preventing the formation of mitochondrial membrane transition pores and reducing cytochrome c release.Citrate produced by the TCA cycle can be transported from mitochondria to cytosol.Cytosolic citrate is used in a number of reactions to produce acetyl-CoA, oxaloacetate and isocitrate.These metabolites are important for lipid synthesis, NAPDH production, and many other central cellular processes.Mitochondrial biogenesis is also a striking feature of cancer metabolic reprogramming.Mitochondria are not only the energy generators but also the factories for synthesizing many essential metabolites for cancer growth, proliferation and metastasis.In addition, the metabolic lactate-based symbiosis is another remarkable characteristic of cancer metabolism.Cancer cells frequently upregulate LDHA to facilitate the conversion of pyruvate to lactate.Lactate is then secreted to tumor microenvironment via MCT4 transporters and can be taken by neighboring cancer cell thanks to MCT1 importers.Lactate is thereafter used for other metabolic pathways in tumors.This metabolic symbiosis facilitates the survival of cancer cells in harsh conditions.Thus, metabolic reprogramming is a major cancer hallmark.It is characterized by the upregulation of a number of inter-connected metabolic pathways providing cancer cells with vital energy and metabolites.This metabolic plasticity is essentially important because it allows cancer cells to effectively and rapidly adapt to the rapidly changing conditions of tumor microenvironment.In addition, the flexibility of cancer bioenergetics also enables rapid proliferation, continuous growth, invasion, metastasis and resistance to anti-cancer therapies.Therefore, further knowledge about cancer metabolic reprogramming is very important for successful development of precise and efficacious anti-cancer metabolism therapies.Dashed arrows indicate indirect effects or multi-step processes.Abbreviations: HK2, hexokinase 2; LDHA, lactate dehydrogenase A; G6PD, glucose-6-phosphate dehydrogenase; 6PGLD, 6-phosphogluconate dehydrogenase; ACC, acetyl-CoA carboxylase; ACLY, ATP citrate lyase; FASN: fatty acid synthase, SCD, stearoyl-CoA desaturase; CPT, carnitine palmitoyltransferase; CPT1C, carnitine palmitoyltransferase 1C; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; UCP, uncoupling proteins; MCT, monocarboxylic acid transporter; ME1, malic enzyme; IDH1, isocitrate dehydrogenase1; GLS1, glutaminase; GLUD,glutamate dehydrogenase; FAO, fatty acid oxidation; ETC, electron transport chain; PPP, pentose phosphate pathway; TCA, tricarboxylic acid cycle; α-KG, alphaketoglutarate.
However, the efficacy of anti-cancer metabolism therapies will need to be carefully evaluated because cancer cells are well known for their metabolic plasticity and heterogeneity1,2,11,21,119,184.That may enable tumors to bypass certain inhibition mediated by therapeutic agents.Furthermore, as we have seen during the past decades, inhibiting individual enzymes or blocking single pathways seldom leads to effective cancer treatment.Therefore,it is highly likely that anti-cancer metabolism approaches need to be combined with other therapies to improve therapeutic effects and clinical outcomes.Further understanding about cancer metabolic reprogramming is certainly needed for effective therapy development.Nevertheless, exploiting the unique features and weakness of tumor metabolism for cancer treatment,detection and monitoring is clearly a very promising direction.
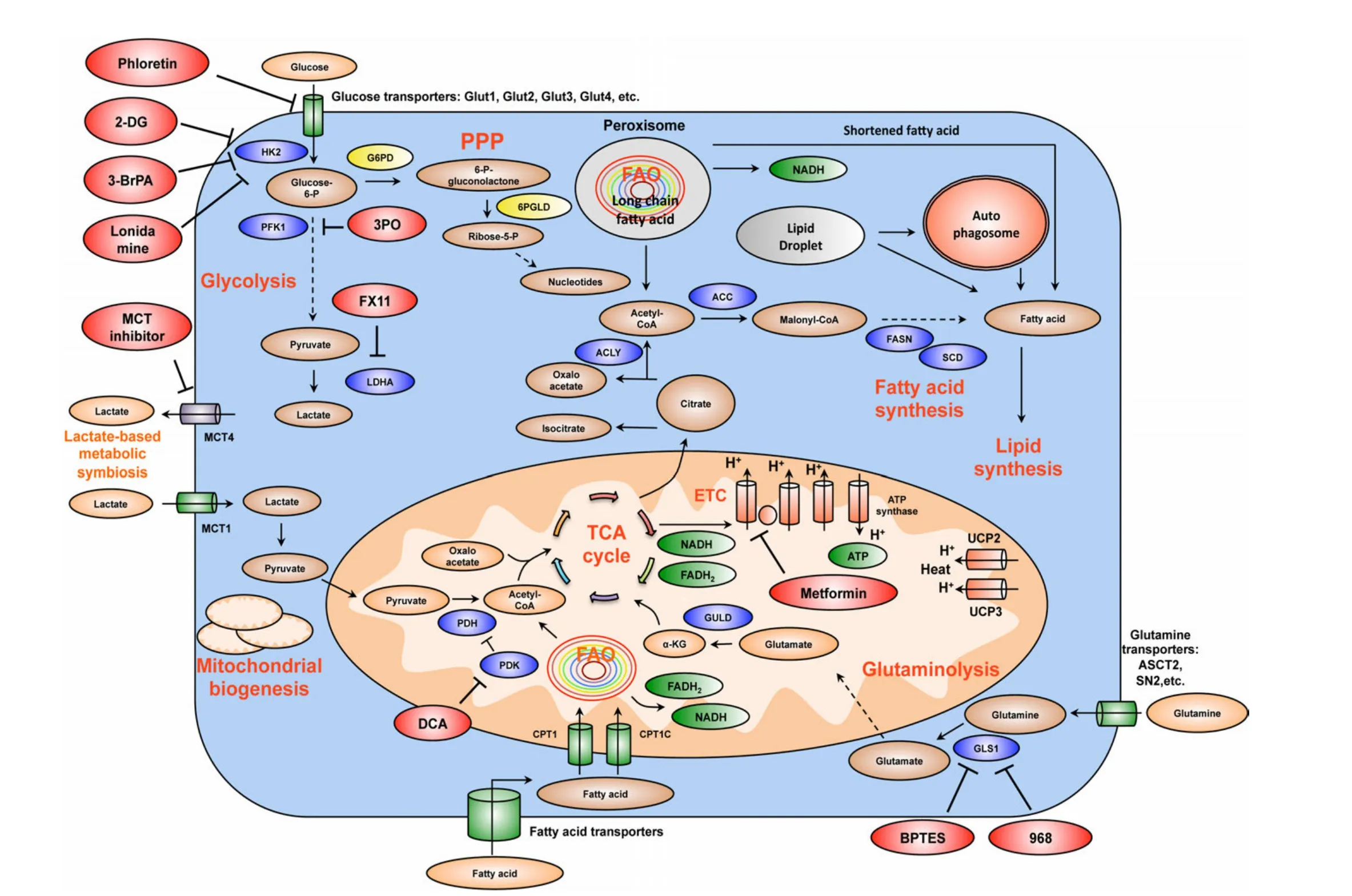
Figure 3 Summary of the mechanism of several important drug candidates for anti-cancer metabolism therapies.Phloretin inhibits the import of glucose, a major source of nutrient for cancer cells.2DG, 3BrPA, and Lonidamine inhibit HK2, a rate-limiting step of glycolytic pathway.3PO blocks PFK1 activation by inhibiting PFKFB3 (PFK2).FX11 selectively inhibits LDHA, a major metabolic enzyme of cancer.BPTES and 968 suppress the function of GLS1.GLS1 is a glutaminolytic enzyme that is highly and selectively upregulated in cancer.DCA inactivates PDH kinase(PDK), thereby increasing PDH activity and enhances the conversion of pyruvate to acetyl-CoA and decreases cancer glycolysis.Metformin blocks energy production of cancer cells by inhibiting mitochondrial complex I, suppresses lipid and protein synthesis, modulates glycolysis.At the organism level, by lowering blood glucose concentration, metformin decreases glucose supply, as well as insulin and insulin-like growth factor signaling availability for tumor cells.MCT inhibitors impair the metabolic lactate-based symbiosis of cancer cells.Many other anticancer metabolism compounds are under development.Targeting cancer metabolism is a very promising direction for anti-cancer therapies.It is expected that inhibitors of tumor metabolism will play an important role in clinical oncology within five or ten years.These medications could be used alone or in combination with other current anti-cancer therapies to increase efficacy.Abbreviations: 2DG, 2-deoxyglucose;3BrPA, 3-bromopyruvate; HK2, hexokinase 2; PFK1, phosphofructose kinase 1; LDHA, lactate dehydrogenase A; GLS1, glutaminase 1; DCA,dicholoroacetate; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; MCT, monocarboxylic acid transporter.
Acknowledgements
Our research was supported by the National Institutes of Health through The University of Texas MD Anderson Cancer Center’s Support Grant CA016672, National Cancer Institute grant RO1CA 089266 (MHL), Directed Medical Research Programs Department of Defense Synergistic Idea Development Award BC062166 (SCY, MHL), the Susan G.Komen Breast Cancer Research Foundation Promise Grant KG081048 (SCY, MHL).LMP is supported by Vietnam Education Foundation, Rosalie B.Hite Foundation and then by Department of Defense Breast Cancer Research Program (Award # W81XWH-10-0171).We apologize for not being able to include all original studies in this review due to space limitation.
Conflict of interest statement
No potential conflicts of interest are disclosed.
1.Hanahan D, Weinberg RA.Hallmarks of cancer: the next generation.Cell 2011;144:646-674.
2.Yeung SJ, Pan J, Lee MH.Roles of p53, MYC and HIF-1 in regulating glycolysis - the seventh hallmark of cancer.Cell Mol Life Sci 2008;65:3981-3999.
3.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB.The biology of cancer: metabolic reprogramming fuels cell growth and proliferation.Cell Metab 2008;7:11-20.
4.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M,Wehrli S, et al.Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis.Proc Natl Acad Sci U S A 2007;104:19345-19350.
5.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB.Brick by brick: metabolism and tumor cell growth.Curr Opin Genet Dev 2008;18:54-61.
6.Thompson CB, Bauer DE, Lum JJ, Hatzivassiliou G, Zong WX, Zhao F, et al.How do cancer cells acquire the fuel needed to support cell growth? Cold Spring Harb Symp Quant Biol 2005;70:357-362.
7.Vander Heiden MG, Cantley LC, Thompson CB.Understanding the Warburg effect: the metabolic requirements of cell proliferation.Science 2009;324:1029-1033.
8.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH,Thompson CB.Growth factors can influence cell growth and survival through effects on glucose metabolism.Mol Cell Biol 2001;21:5899-5912.
9.Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY,Pfeiffer HK, et al.Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction.Proc Natl Acad Sci U S A 2008;105:18782-18787.
10.Wise DR, Thompson CB.Glutamine addiction: a new therapeutic target in cancer.Trends Biochem Sci 2010;35:427-433.
11.Dang CV.c-Myc target genes involved in cell growth, apoptosis,and metabolism.Mol Cell Biol 1999;19:1-11.
12.Dang CV.MYC, microRNAs and glutamine addiction in cancers.Cell Cycle 2009;8:3243-3245.
13.Dang CV.Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle 2010;9:3884-3886.
14.Dang CV.Rethinking the Warburg effect with Myc micromanaging glutamine metabolism.Cancer Res 2010;70:859-862.
15.Dang CV, Le A, Gao P.MYC-induced cancer cell energy metabolism and therapeutic opportunities.Clin Cancer Res 2009;15:6479-6483.
16.Dang CV, Semenza GL.Oncogenic alterations of metabolism.Trends Biochem Sci 1999;24:68-72.
17.Ward PS, Thompson CB.Metabolic reprogramming: a cancer hallmark even warburg did not anticipate.Cancer Cell 2012;21:297-308.
18.Warburg O, Posener K, Negelein E.Ueber den Stoffwechsel der Tumoren.Biochemische Zeitschrift 1924;152:319-344 (German).Reprinted in English in the book On metabolism of tumors by O.Warburg, Publisher: Constable, London, 1930.
19.Warburg O.On respiratory impairment in cancer cells.Science 1956;124:269-270.
20.Warburg O.On the origin of cancer cells.Science 1956;123:309-314.
21.Dang CV.The interplay between MYC and HIF in the Warburg effect.Ernst Schering Found Symp Proc 2007;4:35-53.
22.DeBerardinis RJ, Cheng T.Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer.Oncogene 2010;29:313-324.
23.Hsu PP, Sabatini DM.Cancer cell metabolism: Warburg and beyond.Cell 2008;134:703-707.
24.Jones RG, Thompson CB.Tumor suppressors and cell metabolism:a recipe for cancer growth.Genes Dev 2009;23:537-548.
25.Dang CV, Lewis BC, Dolde C, Dang G, Shim H.Oncogenes in tumor metabolism, tumorigenesis, and apoptosis.J Bioenerg Biomembr 1997;29:345-354.
26.Denko NC.Hypoxia, HIF1 and glucose metabolism in the solid tumour.Nat Rev Cancer 2008;8:705-713.
27.Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E.The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression.Cancer Res 2004;64:2627-2633.
28.Kawauchi K, Araki K, Tobiume K, Tanaka N.p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation.Nat Cell Biol 2008;10:611-618.
29.Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al.TIGAR, a p53-inducible regulator of glycolysis and apoptosis.Cell 2006;126:107-120.
30.Bensaad K, Vousden KH.p53: new roles in metabolism.Trends Cell Biol 2007;17:286-291.
31.Draoui N, Feron O.Lactate shuttles at a glance: from physiological paradigms to anti-cancer treatments.Dis Model Mech 2011;4:727-732.
32.Kennedy KM, Dewhirst MW.Tumor metabolism of lactate:the influence and therapeutic potential for MCT and CD147 regulation.Future Oncol 2010;6:127-148.
33.Semenza GL.Tumor metabolism: cancer cells give and take lactate.J Clin Invest 2008;118:3835-3837.
34.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al.Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism.Cell Cycle 2010;9:3506-3514.
35.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z,Pavlides S, et al.Ketones and lactate increase cancer cell “stemness,”driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics.Cell Cycle 2011;10:1271-1286.
36.Semenza GL, Artemov D, Bedi A, Bhujwalla Z, Chiles K, Feldser D, et al.‘The metabolism of tumours’: 70 years later.Novartis Found Symp 2001;240:251-260; discussion 260-4.
37.Shaw RJ.Glucose metabolism and cancer.Curr Opin Cell Biol 2006;18:598-608.
38.Kim JW, Dang CV.Multifaceted roles of glycolytic enzymes.Trends Biochem Sci 2005;30:142-150.
39.Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G,Fokt I, et al.Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells.Cancer 2011;117:2926-2938.
40.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al.Bidirectional transport of amino acids regulates mTOR and autophagy.Cell 2009;136:521-534.
41.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al.c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism.Nature 2009;458:762-765.
42.Gangrade A, Calin GA.MicroRNAs and Cancer Hallmarks.Cancer Hallmarks 2013;1:50-57.
43.Meng M, Chen S, Lao T, Liang D, Sang N.Nitrogen anabolism underlies the importance of glutaminolysis in proliferating cells.Cell Cycle 2010;9:3921-3932.
44.Riganti C, Gazzano E, Polimeni M, Aldieri E, Ghigo D.The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate.Free Radic Biol Med 2012;53:421-436.
45.Jonas SK, Benedetto C, Flatman A, Hammond RH, Micheletti L, Riley C, et al.Increased activity of 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase in purified cell suspensions and single cells from the uterine cervix in cervical intraepithelial neoplasia.Br J Cancer 1992;66:185-191.
46.Hartmannsberger D, Mack B, Eggert C, Denzel S, Stepp H, Betz CS, et al.Transketolase-like protein 1 confers resistance to serum withdrawal in vitro.Cancer Lett 2011;300:20-29.
47.Vizán P, Alcarraz-Vizán G, Díaz-Moralli S, Solovjeva ON, Frederiks WM, Cascante M.Modulation of pentose phosphate pathway during cell cycle progression in human colon adenocarcinoma cell line HT29.Int J Cancer 2009;124:2789-2796.
48.Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al.p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase.Nat Cell Biol 2011;13:310-316.
49.Gao L, Mejías R, Echevarría M, López-Barneo J.Induction of the glucose-6-phosphate dehydrogenase gene expression by chronic hypoxia in PC12 cells.FEBS Lett 2004;569:256-260.
50.Galtieri A, Tellone E, Ficarra S, Russo A, Bellocco E, Barreca D, et al.Resveratrol treatment induces redox stress in red blood cells: a possible role of caspase 3 in metabolism and anion transport.Biol Chem 2010;391:1057-1065.
51.Boros LG, Torday JS, Lim S, Bassilian S, Cascante M, Lee WN.Transforming growth factor beta2 promotes glucose carbon incorporation into nucleic acid ribose through the nonoxidative pentose cycle in lung epithelial carcinoma cells.Cancer Res 2000;60:1183-1185.
52.Cascante M, Centelles JJ, Veech RL, Lee WN, Boros LG.Role of thiamin (vitamin B-1) and transketolase in tumor cell proliferation.Nutr Cancer 2000;36:150-154.
53.Langbein S, Zerilli M, Zur Hausen A, Staiger W, Rensch-Boschert K, Lukan N, et al.Expression of transketolase TKTL1 predicts colon and urothelial cancer patient survival: Warburg effect reinterpreted.Br J Cancer 2006;94:578-585.
54.Porter SN, Howarth GS, Butler RN.Non-steroidal antiinflammatory drugs and apoptosis in the gastrointestinal tract:potential role of the pentose phosphate pathways.Eur J Pharmacol 2000;397:1-9.
55.Tian WN, Braunstein LD, Apse K, Pang J, Rose M, Tian X, et al.Importance of glucose-6-phosphate dehydrogenase activity in cell death.Am J Physiol 1999;276:C1121-1131.
56.Li D, Zhu Y, Tang Q, Lu H, Li H, Yang Y, et al.A new G6PD knockdown tumor-cell line with reduced proliferation and increased susceptibility to oxidative stress.Cancer Biother Radiopharm 2009;24:81-90.
57.Fico A, Paglialunga F, Cigliano L, Abrescia P, Verde P, Martini G,et al.Glucose-6-phosphate dehydrogenase plays a crucial role in protection from redox-stress-induced apoptosis.Cell Death Differ 2004;11:823-831.
58.Pias EK, Aw TY.Apoptosis in mitotic competent undifferentiated cells is induced by cellular redox imbalance independent of reactive oxygen species production.FASEB J 2002;16:781-790.
59.Cosentino C, Grieco D, Costanzo V.ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair.EMBO J 2011;30:546-555.
60.Efferth T, Fabry U, Osieka R.DNA damage and apoptosis in mononuclear cells from glucose-6-phosphate dehydrogenasede ficient patients (G6PD Aachen variant) after UV irradiation.J Leukoc Biol 2001;69:340-342.
61.Finn PF, Mesires NT, Vine M, Dice JF.Effects of small molecules on chaperone-mediated autophagy.Autophagy 2005;1:141-145.
62.Leopold JA, Walker J, Scribner AW, Voetsch B, Zhang YY, Loscalzo AJ, et al.Glucose-6-phosphate dehydrogenase modulates vascular endothelial growth factor-mediated angiogenesis.J Biol Chem 2003;278:32100-6.
63.Pan S, World CJ, Kovacs CJ, Berk BC.Glucose 6-phosphate dehydrogenase is regulated through c-Src-mediated tyrosine phosphorylation in endothelial cells.Arterioscler Thromb Vasc Biol 2009;29:895-901.
64.Polimeni M, Voena C, Kopecka J, Riganti C, Pescarmona G, Bosia A, et al.Modulation of doxorubicin resistance by the glucose-6-phosphate dehydrogenase activity.Biochem J 2011;439:141-149.
65.Wallace DC.Mitochondria and cancer.Nat Rev Cancer 2012;12:685-698.
66.Cavalli LR, Varella-Garcia M, Liang BC.Diminished tumorigenic phenotype after depletion of mitochondrial DNA.Cell Growth Differ 1997;8:1189-1198.
67.Desjardins P, Frost E, Morais R.Ethidium bromide-induced loss of mitochondrial DNA from primary chicken embryo fibroblasts.Mol Cell Biol 1985;5:1163-1169.
68.King MP, Attardi G.Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation.Science 1989;246:500-503.
69.Magda D, Lecane P, Prescott J, Thiemann P, Ma X, Dranchak PK,et al.mtDNA depletion confers specific gene expression pro files in human cells grown in culture and in xenograft.BMC Genomics 2008;9:521.
70.Morais R, Zinkewich-Péotti K, Parent M, Wang H, Babai F,Zollinger M.Tumor-forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA.Cancer Res 1994;54:3889-3896.
71.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J,Lopez M, et al.Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity.Proc Natl Acad Sci U S A 2010;107:8788-8793.
72.Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, et al.Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis.Mol Cell Biol 2005;25:6225-6234.
73.Santos CR, Schulze A.Lipid metabolism in cancer.FEBS J 2012;279:2610-2623.
74.Medes G, Thomas A, weinhouse S.Metabolism of neoplastic tissue.IV.A study of lipid synthesis in neoplastic tissue slices in vitro.Cancer Res 1953;13:27-29.
75.Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, et al.Fatty acid synthesis: a potential selective target for antineoplastic therapy.Proc Natl Acad Sci U S A 1994;91:6379-6383.
76.Menendez JA, Lupu R.Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis.Nat Rev Cancer 2007;7:763-777.
77.Li JN, Mahmoud MA, Han WF, Ripple M, Pizer ES.Sterol regulatory element-binding protein-1 participates in the regulation of fatty acid synthase expression in colorectal neoplasia.Exp Cell Res 2000;261:159-165.
78.Swinnen JV, Vanderhoydonc F, Elgamal AA, Eelen M, Vercaeren I, Joniau S, et al.Selective activation of the fatty acid synthesis pathway in human prostate cancer.Int J Cancer 2000;88:176-179.
79.Yoon S, Lee MY, Park SW, Moon JS, Koh YK, Ahn YH, et al.Upregulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells.J Biol Chem 2007;282:26122-26131.
80.Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, et al.Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1.Cancer Res 2008;68:1003-1011.
81.Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB.ATP citrate lyase is an important component of cell growth and transformation.Oncogene 2005;24:6314-6322.
82.Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN,Dhanak D, et al.ATP citrate lyase inhibition can suppress tumor cell growth.Cancer Cell 2005;8:311-321.
83.Beckers A, Organe S, Timmermans L, Scheys K, Peeters A,Brusselmans K, et al.Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells.Cancer Res 2007;67:8180-8187.
84.Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N,Robbs BK, Oliveira SS, et al.Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells.Cancer Res 2008;68:1732-1740.
85.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG.Post-translational modi fications and regulation of the RAS superfamily of GTPases as anticancer targets.Nat Rev Drug Discov 2007;6:541-555.
86.Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S,Khosravi F, Trentin GA, et al.Dysregulation of the mevalonate pathway promotes transformation.Proc Natl Acad Sci U S A 2010;107:15051-15056.
87.Kornblau SM, Banker DE, Stirewalt D, Shen D, Lemker E,Verstovsek S, et al.Blockade of adaptive defensive changes in cholesterol uptake and synthesis in AML by the addition of pravastatin to idarubicin + high-dose Ara-C: a phase 1 study.Blood 2007;109:2999-3006.
88.Graf H, Jüngst C, Straub G, Dogan S, Hoffmann RT, Jakobs T,et al.Chemoembolization combined with pravastatin improves survival in patients with hepatocellular carcinoma.Digestion 2008;78:34-38.
89.Kodach LL, Jacobs RJ, Voorneveld PW, Wildenberg ME, Verspaget HW, van Wezel T, et al.Statins augment the chemosensitivity of colorectal cancer cells inducing epigenetic reprogramming and reducing colorectal cancer cell ‘stemness’ via the bone morphogenetic protein pathway.Gut 2011;60:1544-1553.
90.Horton JD.Sterol regulatory element-binding proteins:transcriptional activators of lipid synthesis.Biochem Soc Trans 2002;30:1091-1095.
91.Hardie DG.AMP-activated/SNF1 protein kinases:conserved guardians of cellular energy.Nat Rev Mol Cell Biol 2007;8:774-785.
92.Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, et al.Rb Regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation.Cancer Cell 2009;15:255-269.
93.Freed-Pastor WA, Mizuno H, Zhao X, Langer?d A, Moon SH,Rodriguez-Barrueco R, et al.Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway.Cell 2012;148:244-258.
94.Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D,et al.An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway.Cancer Discov 2011;1:442-456.
95.Samuel VT, Shulman GI.Mechanisms for insulin resistance:common threads and missing links.Cell 2012;148:852-871.
96.Renehan AG, Frystyk J, Flyvbjerg A.Obesity and cancer risk:the role of the insulin-IGF axis.Trends Endocrinol Metab 2006;17:328-336.
97.Rosenzweig SA, Atreya HS.De fining the pathway to insulin-like growth factor system targeting in cancer.Biochem Pharmacol 2010;80:1115-1124.
98.Shoelson SE, Lee J, Gold fine AB.In flammation and insulin resistance.J Clin Invest 2006;116:1793-1801.
99.Kalaany NY, Sabatini DM.Tumours with PI3K activation are resistant to dietary restriction.Nature 2009;458:725-731.
100.Sell Ch.Caloric restriction and insulin-like growth factors in aging and cancer.Horm Metab Res 2003;35:705-711.
101.Carracedo A, Cantley LC, Pandol fiPP.Cancer metabolism: fatty acid oxidation in the limelight.Nat Rev Cancer 2013;13:227-232.
102.Singh R, Cuervo AM.Lipophagy: connecting autophagy and lipid metabolism.Int J Cell Biol 2012;2012:282041.
103.Chiarugi A, D?lle C, Felici R, Ziegler M.The NAD metabolome--a key determinant of cancer cell biology.Nat Rev Cancer 2012;12:741-752.
104.Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M.Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells.Biochim Biophys Acta 2011;1807:726-734.
105.Hosokawa K, Arai F, Yoshihara H, Nakamura Y, Gomei Y,Iwasaki H, et al.Function of oxidative stress in the regulation of hematopoietic stem cell-niche interaction.Biochem Biophys Res Commun 2007;363:578-583.
106.Ito K, Hirao A, Arai F, Takubo K, Matsuoka S, Miyamoto K, et al.Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells.Nat Med 2006;12:446-451.
107.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, et al.Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells.Nature 2004;431:997-1002.
108.Jeon SM, Chandel NS, Hay N.AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress.Nature 2012;485:661-665.
109.Mihaylova MM, Shaw RJ.The AMPK signalling pathway coordinates cell growth, autophagy and metabolism.Nat Cell Biol 2011;13:1016-1023.
110.Diradourian C, Girard J, Pégorier JP.Phosphorylation of PPARs:from molecular characterization to physiological relevance.Biochimie 2005;87:33-38.
111.Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al.Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress.Genes Dev 2011;25:1041-1051.
112.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al.Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment.Nature 2009;461:109-113.
113.Carracedo A, Weiss D, Leliaert AK, Bhasin M, de Boer VC,Laurent G, et al.A metabolic prosurvival role for PML in breast cancer.J Clin Invest 2012;122:3088-3100.
114.Giordano A, Calvani M, Petillo O, Grippo P, Tuccillo F, Melone MA, et al.tBid induces alterations of mitochondrial fatty acid oxidation flux by malonyl-CoA-independent inhibition of carnitine palmitoyltransferase-1.Cell Death Differ 2005;12:603-613.
115.Paumen MB, Ishida Y, Han H, Muramatsu M, Eguchi Y, Tsujimoto Y, et al.Direct interaction of the mitochondrial membrane protein carnitine palmitoyltransferase I with Bcl-2.Biochem Biophys Res Commun 1997;231:523-525.
116.Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M,Korchin B, et al.Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction.J Clin Invest 2010;120:142-156.
117.Vickers AE.Characterization of hepatic mitochondrial injury induced by fatty acid oxidation inhibitors.Toxicol Pathol 2009;37:78-88.
118.Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M.The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation.Cancer Res 2008;68:5198-5205.
119.Hu J, Locasale JW, Bielas JH, O’Sullivan J, Sheahan K, Cantley LC,et al.Heterogeneity of tumor-induced gene expression changes in the human metabolic network.Nat Biotechnol 2013;31:522-529.
120.Kurhanewicz J, Vigneron DB, Brindle K, Chekmenev EY,Comment A, Cunningham CH, et al.Analysis of cancer metabolism by imaging hyperpolarized nuclei: prospects for translation to clinical research.Neoplasia 2011;13:81-97.
121.Nahmias C, Carlson ER, Duncan LD, Blodgett TM, Kennedy J, Long MJ, et al.Positron emission tomography/computerized tomography (PET/CT) scanning for preoperative staging of patients with oral/head and neck cancer.J Oral Maxillofac Surg 2007;65:2524-2535.
122.Blodgett TM, Meltzer CC, Townsend DW.PET/CT: form and function.Radiology 2007;242:360-385.
123.Talbot JN, Gutman F, Fartoux L, Grange JD, Ganne N, Kerrou K,et al.PET/CT in patients with hepatocellular carcinoma using[(18)F] fluorocholine: preliminary comparison with [(18)F]FDG PET/CT.Eur J Nucl Med Mol Imaging 2006;33:1285-1289.
124.Kubota K, Furumoto S, Iwata R, Fukuda H, Kawamura K, Ishiwata K.Comparison of 18F- fluoromethylcholine and 2-deoxy-D-glucose in the distribution of tumor and inflammation.Ann Nucl Med 2006;20:527-533.
125.Breeuwsma AJ, Pruim J, van den Bergh AC, Leliveld AM, Nijman RJ, Dierckx RA, et al.Detection of local, regional, and distant recurrence in patients with psa relapse after external-beam radiotherapy using (11)C-choline positron emission tomography.Int J Radiat Oncol Biol Phys 2010;77:160-164.
126.Piert M, Park H, Khan A, Siddiqui J, Hussain H, Chenevert T, et al.Detection of aggressive primary prostate cancer with 11C-choline PET/CT using multimodality fusion techniques.J Nucl Med 2009;50:1585-1593.
127.Buck AK, Herrmann K, Shen C, Dechow T, Schwaiger M, Wester HJ.Molecular imaging of proliferation in vivo: positron emission tomography with [18F] fluorothymidine.Methods 2009;48:205-215.
128.Buchmann I, Henze M, Engelbrecht S, Eisenhut M, Runz A,Sch?fer M, et al.Comparison of 68Ga-DOTATOC PET and 111In-DTPAOC (Octreoscan) SPECT in patients with neuroendocrine tumours.Eur J Nucl Med Mol Imaging 2007;34:1617-1626.
129.Zhao Z, Yoshida Y, Kurokawa T, Kiyono Y, Mori T, Okazawa H.18F-FES and 18F-FDG PET for differential diagnosis and quantitative evaluation of mesenchymal uterine tumors:correlation with immunohistochemical analysis.J Nucl Med 2013;54:499-506.
130.Haubner R.Alphavbeta3-integrin imaging: a new approach to characterise angiogenesis? Eur J Nucl Med Mol Imaging 2006;33 Suppl 1:54-63.
131.Wagner B, Anton M, Nekolla SG, Reder S, Henke J, Seidl S,et al.Noninvasive characterization of myocardial molecular interventions by integrated positron emission tomography and computed tomography.J Am Coll Cardiol 2006;48:2107-2115.
132.Chuang CF, Chan AA, Larson D, Verhey LJ, McDermott M,Nelson SJ, et al.Potential value of MR spectroscopic imaging for the radiosurgical management of patients with recurrent highgrade gliomas.Technol Cancer Res Treat 2007;6:375-382.
133.Kurhanewicz J, Vigneron DB.Advances in MR spectroscopy of the prostate.Magn Reson Imaging Clin N Am 2008;16:697-710, ix-x.
134.Mountford C, Ramadan S, Stanwell P, Malycha P.Proton MRS of the breast in the clinical setting.NMR Biomed 2009;22:54-64.
135.Geraghty PR, van den Bosch MA, Spielman DM, Hunjan S,Birdwell RL, Fong KJ, et al.MRI and (1)H MRS of the breast:presence of a choline peak as malignancy marker is related to K21 value of the tumor in patients with invasive ductal carcinoma.Breast J 2008;14:574-580.
136.Glunde K, Ackerstaff E, Mori N, Jacobs MA, Bhujwalla ZM.Choline phospholipid metabolism in cancer: consequences for molecular pharmaceutical interventions.Mol Pharm 2006;3:496-506.
137.Haddadin IS, McIntosh A, Meisamy S, Corum C, Styczynski Snyder AL, Powell NJ, et al.Metabolite quantification and highfield MRS in breast cancer.NMR Biomed 2009;22:65-76.
138.Nelson SJ, Graves E, Pirzkall A, Li X, Antiniw Chan A, Vigneron DB, et al.In vivo molecular imaging for planning radiation therapy of gliomas: an application of 1H MRSI.J Magn Reson Imaging 2002;16:464-476.
139.Gillies RJ, Morse DL.In vivo magnetic resonance spectroscopy in cancer.Annu Rev Biomed Eng 2005;7:287-326.
140.Chen J, Huang SL, Li T, Chen XL.In vivo research in astrocytoma cell proliferation with 1H-magnetic resonance spectroscopy:correlation with histopathology and immunohistochemistry.Neuroradiology 2006;48:312-318.
141.Magalhaes A, Godfrey W, Shen Y, Hu J, Smith W.Proton magnetic resonance spectroscopy of brain tumors correlated with pathology.Acad Radiol 2005;12:51-57.
142.McKnight TR, Lamborn KR, Love TD, Berger MS, Chang S,Dillon WP, et al.Correlation of magnetic resonance spectroscopic and growth characteristics within Grades II and III gliomas.J Neurosurg 2007;106:660-666.
143.Spampinato MV, Smith JK, Kwock L, Ewend M, Grimme JD,Camacho DL, et al.Cerebral blood volume measurements and proton MR spectroscopy in grading of oligodendroglial tumors.AJR Am J Roentgenol 2007;188:204-212.
144.Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, Ferrone M, et al.Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate.Sci Transl Med 2013;5:198ra108.
145.Sardanelli F, Fausto A, Podo F.MR spectroscopy of the breast.Radiol Med 2008;113:56-64.
146.Kwock L, Smith JK, Castillo M, Ewend MG, Cush S, Hensing T, et al.Clinical applications of proton MR spectroscopy in oncology.Technol Cancer Res Treat 2002;1:17-28.
147.DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, et al.Fulllength human glutaminase in complex with an allosteric inhibitor.Biochemistry 2011;50:10764-10770.
148.Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al.Targeting mitochondrial glutaminase activity inhibits oncogenic transformation.Cancer Cell 2010;18:207-219.
149.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al.Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells.Cell Metab 2012;15:110-121.
150.Jiang JK, Boxer MB, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, et al.Evaluation of thieno[3,2-b]pyrrole[3,2-d]pyridazinones as activators of the tumor cell specific M2 isoform of pyruvate kinase.Bioorg Med Chem Lett 2010;20:3387-3393.
151.Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM,Bellinger G, et al.PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells.Cell 2013;155:397-409.
152.Zhou W, Simpson PJ, McFadden JM, et al.Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells.Cancer Res 2003;63:7330-7337.
153.Brahimi-Horn MC, Bellot G, Pouysségur J.Hypoxia and energetic tumour metabolism.Curr Opin Genet Dev 2011;21:67-72.
154.Morris JC, Chiche J, Grellier C, Lopez M, Bornaghi LF, Maresca A, et al.Targeting hypoxic tumor cell viability with carbohydratebased carbonic anhydrase IX and XII inhibitors.J Med Chem 2011;54:6905-6918.
155.Semenza GL.HIF-1: upstream and downstream of cancer metabolism.Curr Opin Genet Dev 2010;20:51-56.
156.Semenza GL.Hypoxia-inducible factors in physiology and medicine.Cell 2012;148:399-408.
157.Fang J, Quinones QJ, Holman TL, Morowitz MJ, Wang Q,Zhao H, et al.The H+-linked monocarboxylate transporter(MCT1/SLC16A1): a potential therapeutic target for high-risk neuroblastoma.Mol Pharmacol 2006;70:2108-2115.
158.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al.Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice.J Clin Invest 2008;118:3930-3942.
159.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J,Edinger M, et al.Inhibitory effect of tumor cell-derived lactic acid on human T cells.Blood 2007;109:3812-3819.
160.Izumi H, Takahashi M, Uramoto H, Nakayama Y, Oyama T, Wang KY, et al.Monocarboxylate transporters 1 and 4 are involved in the invasion activity of human lung cancer cells.Cancer Sci 2011;102:1007-1013.
161.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al.Metabolic modulation of glioblastoma with dichloroacetate.Sci Transl Med 2010;2:31ra34.
162.Papandreou I, Goliasova T, Denko NC.Anticancer drugs that target metabolism: Is dichloroacetate the new paradigm? Int J Cancer 2011;128:1001-1008.
163.Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C,Thompson R, et al.A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth.Cancer Cell 2007;11:37-51.
164.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM,et al.Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression.Proc Natl Acad Sci U S A 2010;107:2037-2042.
165.Maher JC, Wangpaichitr M, Savaraj N, Kurtoglu M, Lampidis TJ.Hypoxia-inducible factor-1 confers resistance to the glycolytic inhibitor 2-deoxy-D-glucose.Mol Cancer Ther 2007;6:732-741.
166.Pelicano H, Martin DS, Xu RH, Huang P.Glycolysis inhibition for anticancer treatment.Oncogene 2006;25:4633-4646.
167.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, DepinhoRA, et al.The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin.Science 2005;310:1642-1646.
168.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD.Metformin and reduced risk of cancer in diabetic patients.BMJ 2005;330:1304-1305.
169.Bowker SL, Majumdar SR, Veugelers P, Johnson JA.Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: Response to Farooki and Schneider.Diabetes Care 2006;29:1990-1991.
170.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K.Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission.Cancer Res 2009;69:7507-7511.
171.Dahabreh IJ, Linardou H, Siannis F, Kosmidis P, Bafaloukos D, Murray S.Somatic EGFR mutation and gene copy gain as predictive biomarkers for response to tyrosine kinase inhibitors in non-small cell lung cancer.Clin Cancer Res 2010;16:291-303.
172.Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E,Ciardiello F.Implications for KRAS status and EGFR-targeted therapies in metastatic CRC.Nat Rev Clin Oncol 2009;6:519-527.
173.Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y.De ficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells.J Cell Biol 2007;178:93-105.
174.Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H,et al.Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells.Science 2009;325:1555-1559.
175.Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, et al.Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth.Mol Cancer Ther 2008;7:110-120.
176.Derst C, Henseling J, R?hm KH.Engineering the substrate specificity of Escherichia coli asparaginase.II.Selective reduction of glutaminase activity by amino acid replacements at position 248.Protein Sci 2000;9:2009-2017.
177.Bach SJ, Swaine D.The effect of arginase on the retardation of tumour growth.Br J Cancer 1965;19:379-386.
178.Ni Y, Schwaneberg U, Sun ZH.Arginine deiminase, a potential anti-tumor drug.Cancer Lett 2008;261:1-11.
179.Neuman RE, Mccoy TA.Dual requirement of Walker carcinosarcoma 256 in vitro for asparagine and glutamine.Science 1956;124:124-125.
180.Ardawi MS, Newsholme EA.Glutamine metabolism in lymphocytes of the rat.Biochem J 1983;212:835-842.
181.Bruzzone S, Fruscione F, Morando S, Ferrando T, Poggi A, Garuti A, et al.Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE.PLoS One 2009;4:e7897.
182.Holen K, Saltz LB, Hollywood E, Burk K, Hanauske AR.The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor.Invest New Drugs 2008;26:45-51.
183.Wen YY, Chou PC, Phan L, Su CH, Chen J, Hsieh YC, Xue YW, et al.DNA Damage-Mediated c-Myc Degradation Requires 14-3-3 Sigma.Cancer Hallmarks 2013;1:3-17.
184.Dang CV.Links between metabolism and cancer.Genes Dev 2012;26:877-890.
 Cancer Biology & Medicine2014年1期
Cancer Biology & Medicine2014年1期
- Cancer Biology & Medicine的其它文章
- Inhalation treatment of lung cancer: the influence of composition, size and shape of nanocarriers on their lung accumulation and retention
- Interferon-alpha-2b induces autophagy in hepatocellular carcinoma cells through Beclin1 pathway
- Uptake of prostate cancer screening and associated factors among Chinese men aged 50 or more: a population-based survey
- pH-responsive mesoporous silica nanoparticles employed in controlled drug delivery systems for cancer treatment
- Mechanistic considerations for the use of monoclonal antibodies for cancer therapy
- Instructions for Authors