
Systemic treatment in EGFR-ALK NSCLC patients: second line therapy and beyond
2014-03-29 05:58:13NikiKarachaliouRafaelRosell
Cancer Biology & Medicine 2014年3期
Niki Karachaliou, Rafael Rosell,2,3
1Translational Research Unit, Dr Rosell Oncology Institute, Quirón Dexeus University Hospital, 08028 Barcelona, Spain;
2Cancer Biology and Precision Medicine Program, Catalan Institute of Oncology, Hospital Germans Trias i Pujol, Ctra Canyet s/n, 08916 Badalona, Barcelona, Spain;
3Fundación Molecular Oncology Research (MORe), Sabino Arana 5-19, 08028 Barcelona, Spain
Systemic treatment in EGFR-ALK NSCLC patients: second line therapy and beyond
Niki Karachaliou1, Rafael Rosell1,2,3
1Translational Research Unit, Dr Rosell Oncology Institute, Quirón Dexeus University Hospital, 08028 Barcelona, Spain;
2Cancer Biology and Precision Medicine Program, Catalan Institute of Oncology, Hospital Germans Trias i Pujol, Ctra Canyet s/n, 08916 Badalona, Barcelona, Spain;
3Fundación Molecular Oncology Research (MORe), Sabino Arana 5-19, 08028 Barcelona, Spain
Lung cancer is the most frequently diagnosed cancer and a leading cause of cancer mortality worldwide, with adenocarcinoma being the most common histological subtype. Deeper understanding of the pathobiology of non-small cell lung cancer (NSCLC) has led to the development of small molecules that target genetic mutations known to play critical roles in progression to metastatic disease and to in fl uence response to targeted therapies.e principle goal of precision medicine is to de fi ne those patient populations most likely to respond to targeted therapies. However, the cancer genome landscape is composed of relatively few “mountains” [representing the most commonly mutated genes like KS, epidermal growth factor (EGFR), and anaplastic lymphoma kinase (ALK)] and a vast number of “hills” (representing low frequency but potentially actionable mutations). Low-frequency lesions that a ff ect a druggable gene product allow a relatively small population of cancer patients for targeted therapy to be selected.
Lung cancer; epidermal growth factor (EGFR); anaplastic lymphoma kinase fusions (ALK fusions); tyrosine kinase inhibitors (TKIs); TKI resistance
Introduction
In recent years, diagnosis and treatment of patients with advanced lung cancer have undergone transformational changes. The current paradigm for prescribing novel targeted therapies is based on selecting patients according to the presence of specific oncogenic abnormalities in the tumor. The efficacy of therapy targeted at a specific oncogene is convincing evidence of “oncogene addiction”, or the concept that some cancers rely on or are “addicted to” a specific gene for their survival and proliferation1.
The first such abnormalities discovered in lung cancer were epidermal growth factor (EGFR) kinase domain mutations; tumors with these mutations were found to be sensitive to EGFRtyrosine kinase inhibitors (TKIs)2.e echinoderm microtubuleassociated protein-like 4- anaplastic lymphoma kinase (EML4-ALK) fusion has emerged as the second most important driver oncogene in lung cancer and the first targetable fusion oncokinase to be identi fi ed in 4%-6% of lung adenocarcinomas3,4. Crizotinib, an oral small-molecule inhibitor of ALK and c-MET receptor kinases, is now approved for treatment of ALK positive advanced non-small cell lung cancer (NSCLC), based on the results of two pivotal studies5-7.
Among EGFR or ALK mutated NSCLCs, the percentage of complete response is negligible and therefore novel, effective, safe treatments need to be tested and developed. To this end, repurposing an existing drug for treatment of NSCLC is also a worthy goal.
EGFR NSCLC patients: second line therapy and beyond
Our group was the first to evaluate the feasibility of largescale screening for EGFR mutations in patients with advancedNSCLC and analyze the association between the mutations and the outcome of erlotinib treatment8. Since the introduction of erlotinib and gefitinib, patients with metastatic EGFR positive lung cancer can be offered a therapeutic alternative that has proven its superiority over standard platinum-based chemotherapy2,9. In the EURTAC study, in which erlotinib was compared with platinum-doublet chemotherapy as first-line treatment for patients with EGFR-mutant NSCLC, erlotinib demonstrated a signi fi cant improvement in the overall response rate (ORR) and median progression free survival (PFS)2. On the basis of this study, in May 2013 the U.S Food and Drug Administration (FDA) approved erlotinib for use in patients with lung cancers harboring EGFR exon 19 deletions and EGFR L858R substitutions2.
Whereas reversible EGFR TKIs compete with ATP in the kinase domain of EGFR, 2ndgeneration EGFR TKIs, also compete for ATP binding but then covalently bind at the edge of the ATP binding cleft on Cys773 of EGFR via the Michael mechanism (addition of nucleophile to an α, β unsaturated carbonyl)10. Currently there are two lead 2ndgeneration EGFR TKI candidates, afatinib and dacomitinib, that are active against EGFR mutations with acquired resistance to erlotinib or ge fi tinib11. Afatinib exhibits superior anticancer activity in lung cancer patients harboring gefitinib/erlotinib-resistant mutant EGFR (including T790M, exon 20 insertion, and T790M/ L858R double mutation)12. Based on the results of the LUX-Lung 3, the FDA has approved afatinib as a new fi rst-line treatment for patients with metastatic EGFR-mutated NSCLC13. In the pooled analysis from LUX-Lung 3 and 6, presented this year at ASCO, afatinib prolonged survival of lung cancer patients whose tumors have common EGFR mutations by a median of three months compared with standard chemotherapy and signi fi cantly reduced the risk of death by 19%. The most pronounced reduction in risk of death was 41% in patients whose tumors had the most common exon 19 deletion EGFR mutation; for patients with the L8585R mutation there was no impact on overall survival14.
Despite the recent paradigm shiin the treatment of NSCLC patients, with a move toward biomarker-directed therapy, preclinical data has seldom been incorporated into clinical practice, since the existence of multiple resistance mechanisms complicates the selection of optimal biomarkers. Among patients progressing to first generation EGFR TKIs, 50% have tumors with a secondary T790M mutation15. The emergence of the T790M EGFR gatekeeper mutation and up-regulation of downstream signaling by MET amplification have been described as the two main mechanisms responsible for acquired resistance16. However a phase III trial enrolling only patients with MET-positive tumors was stopped in early March 2014 due to futility; there was no evidence to suggest a positive e ff ect of addition of onartuzumab to erlotinib17.
Other mechanisms include EGFR amplifications, PI3KCA mutations or a transition from epithelial to mesenchymal differentiation. For a small percentage of resistant tumors, histological transformation occurs to small cell lung cancer (SCLC)16. We recently reported possible causes of resistance to EGFR TKIs in EGFR-mutant NSCLC patients: the high coexistence of the pretreatment somatic T790M mutation, with a clear impact on PFS, and the role of BIM mRNA expression as an independent prognostic marker18. Pretreatment T790M can be detected in more than 60% of cases18. In fact, using a PCR-PNA assay pretreatment, T790M mutations were detected in 65.26% of patients18. These results reinforce the need for 2ndand 3rdgeneration EGFR TKIs, while taking into account existing data that suggest use of erlotinib or ge fi tinib beyond progression, with added chemotherapy, radiotherapy or best supportive care may improve survival19. Although afatinib and dacomitinib have been introduced to overcome acquired resistance, they showed limited efficacy in NSCLC with T790M and were more than 100-fold less potent in NSCLC cells with EGFR T790M mutation than in NSCLC cells with EGFR activating mutation20.
CO-1686 is a novel covalent inhibitor that irreversibly and selectively targets both the initial activating EGFR mutations and the T790M secondary acquired resistance mutation21. To investigate its use as a single agent, CO-1686 is being evaluated in a phase I/II trial in EGFR-mutant NSCLC patients previously treated with first-line gefitinib or erlotinib (NCT01526928)22. In the phase I study, and based on early findings from the ongoing phase II trial, the agent yielded a 58% ORR across all dose levels in trial participants with biopsy-confirmed EGFR T790M mutations. Additionally, the compound did not cause the skin rash and diarrhea commonly associated with earlier generations of EGFR inhibitors23. AZD9291 showed high activity in preclinical studies and was well tolerated in animal models. Xenograft studies identified a breakdown metabolite of AZD9291 called AZ5104 that is about five times as potent as AZD9291 itself24. In the phase I study of AZD9291 in EGFR mutant patients resistant to standard EGFR TKIs, 50% of patients experienced tumor shrinkage and the drug worked particularly well in patients with the T790M mutation25.
A consistent proportion of EGFR mutant patients, approximately 30%, never respond to EGFR TKIs due to primary resistance. BIM mRNA expression could enrich the molecular diagnosis in patients with EGFR mutations, by identifying patients unlikely to respond and those with stable disease for whom treatment may not be effective due to early adaptive resistance18,26. It is worth mentioning that, besides BIMexpression, a common BIM deletion polymorphism impairs generation of the proapoptotic isoform required for EGFR TKIs27. This polymorphism confers an inherent drug-resistant phenotype that can be circumvented with the addition of the histone deacetylase (HDAC) inhibitor vorinostat to EGFR TKIs28.
The two primary signaling pathways activated by EGFR include the mitogen-activated protein kinase (MAPK), and the phosphoinositide-3-kinase (PI3K), axes. Src tyrosine kinases and activation of the signal transducer and activator of transcription 3 (STAT3) pathways and downstream signaling have also been well documented29. EGFR phosphorylation leads to recruitment of multiple e ff ector proteins through recognition and binding of Src-homology 2 domain-containing phosphatase 2 (SHP2) to phosphotyrosine motifs on the receptor29. SHP2 (encoded by PTPN11) is a ubiquitously expressed SH2 domaincontaining protein tyrosine phosphatase (PTP). Despite its direct function in protein dephosphorylation, SHP2 plays an overall positive role in transducing signals initiated from growth factors/cytokines and extracellular matrix proteins, and initiating various downstream signaling cascades, including the PI3K and MAPK29,30. By contrast, SHP2 functions as a negative regulator of the JAK/STAT pathway31.
In 2004, Sordella and colleagues were able to demonstrate the differential EGF-induced tyrosine phosphorylation pattern seen with wild-type (WT) and mutant EGFR receptors32. For instance, Y845 is highly phosphorylated in the L858R missense mutant, but not in the WT or deletion mutant, and hence appears to be unique in distinguishing between the two types of EGFR mutations32. Y845 (pY845) phosphorylation stabilizes the activation loop, maintains the enzyme in an active state, and regulates STAT3/5 activity32. Surprisingly, the EGFR L858R mutation leads to decreased ability to activate ERK compared to WT EGFR which correlates with decreased EGFR internalization, reduced phosphorylation of SHP2, hyperactivity of STAT3 and reduced sensitivity to gefitinib33. Lazzara and colleagues found that SHP2 Y542 phosphorylation was not induced in response to EGF in the H3255 cells, which harbor the missense L858R exon 21 mutation, suggesting that SHP2 activity may be less efficiently promoted by EGFR L858R and the STAT3 pathway may be more active34. The main problem is that STAT3 activation is not abrogated by single EGFR TKI treatment with ge fi tinib, erlotinib, afatinib or dacomitinib20,34-36. Indeed, the second generation irreversible EGFR TKIs, afatinib or dacomitinib, not only do not abrogate, but may even induce STAT3 phosphorylation in gefitinib or erlotinib resistant cell lines such as H1975 or PC9-R20.
Afatinib activates interleukin-6 receptor (IL-6R)/JAK1/ STAT3 signaling via autocrine IL-6 secretion in both cells. Blockade of IL-6R/JAK1 significantly increased sensitivity to afatinib through inhibition of afatinib-induced STAT3 activation. The role of the paracrine IL-6R/JAK1/STAT3 loop between stroma and cancer cells in the development of drug resistance is crucial20. Yao et al. uncovered the existence of a subpopulation of cells intrinsically resistant to erlotinib which display features suggestive of epithelial-to-mesenchymal transition (EMT) in NSCLC-derived cell lines and early-stage tumors before erlotinib treatment37. Activation of TGF-beta-mediated signaling was sufficient to induce these phenotypes. An increased TGF-beta-dependent IL-6 secretion released previously addicted lung tumor cells from their EGFR dependency.erefore, both tumor cell-autonomous mechanisms and/or activation of the tumor microenvironment could contribute to primary and acquired erlotinib resistance, and as such, treatments based on EGFR inhibition may not be su ffi cient for the e ff ective treatment of lung-cancer patients harboring mutant EGFR37. Combination of EGFR TKIs with drugs that target the IL-6R/JAK1/STAT3 pathway can be a rational synthetic lethal approach to overcome this mechanism of resistance (Figure 1).
Furthermore, tumor cells exposed to reversible or irreversible EGFR TKIs display early resistance dependent on MET-independent activation of BCL-2/BCL-XL survival signaling26. According to the study of Fan and colleagues, such cells displayed a quiescence-like state that was readily reversed aer withdrawal of targeted inhibitors. BCL-2 induction and p-STAT3 (Y705) activation were found within the residual tumor cells surviving the initial antitumor response to targeted therapies26.
Although MAPK signaling is more commonly associated with receptor tyrosine kinases, it has been demonstrated that TGFBR1 also has tyrosine kinase activity. TGFBR1 directly phosphorylates tyrosine on ShcA, allowing for interaction with Grb2 and Sos.is leads to Ras activation, downstream ERK phosphorylation and BIM downregulation38. Furthermore, TGF-b can induce expression of the transcription factor Snail, which leads to repression of E-cadherin transcription. SMAD-independent activation of Snail is a result of RAS/ERK activation. Although the mechanism remains incomplete and deserves further study, in the tumor seing of aberrant TGF-b signaling, TGF-b actively represses E-cadherin and promotes EMT38.
More interesting is the fact that ADAM metallopeptidase domain 17 (ADAM17) or tumor necrosis factor alpha converting enzyme (TACE) targeting of TGFBR1 has been linked to ERK activation in nonprostate cells, resulting in shedding of the TGFBR1 ectodomain38.ere is evidence that active vitamin D (1,25(OH)2D) therapy effectively inhibits ADAM17 and suppresses TGF/EGFR-pathway39. Finally, Yuanet al. recently demonstrated that a novel long noncoding RNA activated by TGF-b (lncRNA-ATB) ampli fi es the prometastatic effect of TGF-b via two independent mechanisms: firstly by inhibiting miR-200s, releasing activity of ZEB1 and ZEB2 and promoting EMT, and secondly by stabilizing IL11-mRNA and increasing autocrine IL-11-STAT3 signaling40,41.
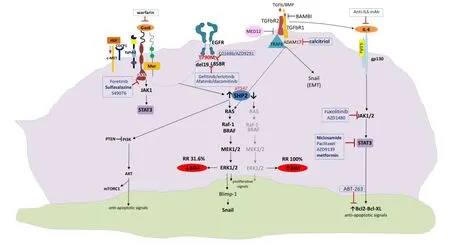
Figure 1 Crosstalk signaling pathways as potential mechanisms of resistance to reversible or irreversible EGFR TKIs. EGFR, epidermal growth factor; TKIs, tyrosine kinase inhibitors.
BIM expression in treatment na?ve cancers predicts responsiveness to EGFR TKIs, but almost 2/3 of patients have low BIM mRNA levels at baseline18. SHP2, which is downstream of both EGFR and several other tyrosine kinase receptors, is required for sustained activation of ERK and BIM downregulation42,43. Upon activation of MET by its ligand, hepatocyte growth factor (HGF) which is provided by stromal cells, EGFR signaling is dramatically altered44. Indeed, HGF enables EGFR to interact with proteins which are known to be markers of a highly metastatic phenotype like the CUB domaincontaining protein-1 (CDCP1), Ephrin Type-A Receptor 2 (EphA2), but mainly AXL and Mer; these interactions cannot be a ff ected by EGFR TKI treatment. Interestingly, STAT3 signaling can be hyperactive due to upstream pathways including not only IL-6 and JAK but also AXL, providing further opportunities for combination therapies (Figure 1).
ALK NSCLC patients: second line therapy and beyond
The novel ALK fusion is formed by a rearrangement occurring on the short arm of chromosome 2 and involves the N-terminal portion of the EML4 protein and the intracellular signaling portion of the ALK tyrosine kinase receptor45. EML4-ALK generates a transforming tyrosine kinase with as many as nine different variants identified46. ALK-dependent mitogenic signaling is largely mediated via theS/MAP kinase pathway as well as ALK-driven PI3K activation.e JAK/STAT3 pathway also provides essential survival signals and modulates cellular metabolism regulating the mitochondrial oxidation chain. STAT3 is activated by ALK, either directly or through JAK47. Crizotinib, an ATP-competitive aminopyridine inhibits tyrosine phosphorylation of ALK with an IC50of 20-40 nM and response in 57% of patients with ALK-rearrangement positive lung cancer5. However, most patients develop resistance to crizotinib, typically within one to two years. Studies of ALK-rearranged lung cancers with acquired resistance to crizotinib have identi fi ed ALK fusion gene ampli fi cation and secondary ALK TK domain mutations (L1196M and G1269A) in about one third of cases48.
The success of a targeted drug is critically dependent on a specific and sensitive screening assay to detect the molecular drug target. The gold standard for detection of predictive ALK rearrangements is currently break-apart fluorescence in situ hybridization (FISH) as it is able to detect all known ALK-rearrangements and was clinically validated in crizotinib clinical trials7. However, the ALK FISH assay is fraught with technical challenges, including FISH signal instability and scoring difficulties. Although ALK FISH is clinically validated, the assay can be technically challenging and other diagnostic modalities are being explored, including immunohistochemistry (IHC) and reverse transcriptase–polymerase chain reaction (RT-PCR). Very interestingly, high frequency of ALK rearrangements in thyroid cancer from atomic bomb survivors in Japan was detected by a highly sensitive RT-PCR assay from archival paraffin blocks but was not confirmed by FISH or other methods49. Targeted resequencing has recently been proved to be a promising method for ALK gene fusion detection in NSCLC, with results correlating signi fi cantly with those from FISH, RT-PCR, and IHC50.
As with the majority of targeted agents, the tremendous excitement and enthusiasm sparked by crizotinib is tempered by the reality that a fraction of the target tumors are refractory from time of treatment initiation and most patients will eventually relapse and develop resistance aer initial response. ALK kinase mutations and ALK fusion copy number gain (CNG) have been termed ALK-dominant mechanisms of resistance since tumors harboring these mechanisms presumably preserve ALK signaling despite the presence of crizotinib and are still dependent on that pathway for survival51. Data from several second-generation ALK inhibitors [ceritinib (LDK378), AP26113, (alectinib)] demonstrate response rates of 55% to 60% in crizotinib-resistant ALK positive NSCLC patients, with observed or predicted disease control rates of approximately 90%51. Ceritinib (LDK378) is an oral, small-molecule, ATP-competitive, ALK TKI52. Ceritinib achieved 56% response rate in patients previously treated with crizotinib with various resistance mutations in ALK; also in patients without detectable mutations52. On April 29th2014, the FDA granted accelerated approval to ceritinib for treatment of patients with ALK positive, metastatic NSCLC with disease progression on or intolerant to crizotinib.e approval of ceritinib was based on the results of a multicenter, single-arm, open-label clinical trial enrolling 163 patients with metastatic ALK-positive NSCLC who had progressed on or were intolerant to crizotinib.is study is part of the ongoing clinical trial program in this patient population. Zykadia achieved an ORR of 54.6% and a median duration of response of 7.4 months based on investigator assessment53. However, while ceritinib is able to effectively suppress many crizotinib-resistant mutations, the G1202R and F1174V/C mutants are resistant to ceritinib48.
One of the questions surrounding second-generation ALK inhibitors is exactly how they will fit into the treatment landscape of ALK positive NSCLC and whether they stand a chance of outshining crizotinib at the frontline. Furthermore, encouraging early clinical results in NSCLC have demonstrated that ganetespib, a novel triazolone inhibitor of HSP90, may offer a potential strategy to target ALK inhibition by inducing substantial antitumor responses and overcoming acquired resistance in patients with ALK positive lung cancer. When combined with ganetespib, crizotinib displayed superior antitumor efficacy compared with monotherapy in H3122 NSCLC xenografts54. Recently, Richards et al. found that sensitivity of EML4-ALK variants to HSP90 inhibitors differs depending on whether the breakpoint of the fusion protein interrupts the globular TAPE (tandem atypical propeller in EMLs) domain. They determined the molecular structure of a conserved, tubulin-binding region of EML1 that reveals an unexpected protein fold55. This region is disrupted in almost 70% of EML4-ALK fusions found in patients, causing them to be sensitive to drugs that target HSP90, a cellular factor that stabilizes misfolded protein. These findings suggest that the truncation of a globular domain at the translocation breakpoint may prove generally predictive of HSP90 inhibitor sensitivity in cancers driven by fusion oncogenes55.
Other “ALK-independent” mechanisms, such as the activation of compensatory signaling pathways, may also confer resistance to targeted ALK agents. Bypass signaling, by v-Kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) or EGFR, has also been described as a mechanism of resistance to crizotinib in ALK positive NSCLC51. EGF-induced activation of EGFR, HER2, and HER3, as well as a reduced level of ALK activation, was associated with sustained downstream signaling in the presence of ALK inhibitors, indicative of a shiin survival dependency from the ALK signaling pathway to HER family pathways in ALK-TKI–resistant cells56.e combination of an ALK inhibitor and an EGFR inhibitor induced apoptosis, further supporting the notion that the EGFR signaling pathway contributes to survival in cells resistant to ALK inhibitors56. ALK rearrangements and EGFR mutations could coexist in a small subgroup of NSCLC. Advanced pulmonary adenocarcinomas with such co-alterations could have diverse responses to EGFRTKIs and crizotinib57. Relative phospho-ALK and phospho-EGFR levels could predict the efficacy of EGFR-TKI and crizotinib. Yang et al. recently described co-altered EGFR and ALK in a large cohort of NSCLC and found that 3.9% of EGFR mutant and 18.6% of ALK rearranged tumors have co-alterations57. Tumors harboring co-altered EGFR and ALK could have diverse responses to first-line EGFR-TKIs, which were associated with phospho-EGFR levels57. Phospho-ALK levels correlated with efficacy of subsequent crizotinib treatment. In clinical practice, we should pay aention to the speci fi c biological behavior and corresponding management of NSCLC with dual altered EGFR and ALK genes57.
ALK and its ligand pleiotrophin are required for self-renewal and tumorigenicity of glioblastoma stem cells (GSCs)58. The pleiotrophin-ALK axis is activated by SOX2 and may be a promising target for therapy in ALK signaling-dependent tumors58. Finally, Takezawa et al. demonstrated that expression of BIM and survivin are independently regulated by ERK and STAT3 signaling pathways, respectively, and are implicated in ALK-TKI-induced apoptosis in NSCLC cells positive for EML4-ALK59. A selective inhibitor of ALK kinase activity (more potent than crizotinib), NVP-TAE68, inhibits STAT3 phosphorylation and downregulates survivin in H2228 cells, but fails to inhibit ERK phosphorylation and upregulate BIM. In a recent study, inhibition of both STAT3 and ERK pathways with the combination of TAE684 and a MEK inhibitor, AZD6244, was associated with a marked increase in the number of apoptotic cells60(Figure 2).
Conclusion
TKIs are effective anti-cancer therapies but resistance to these agents eventually develops. Several models of resistance to TKIs have been studied, including resistance to EGFR inhibitors or ALK inhibitors. A decade aer the discovery of EGFR mutations, treatment is still incomplete and growing evidence indicates that targeting STAT3 or upstream or downstream components could cause induce synthetic lethal e ff ects and achieve beer outcomes. Repurposing drugs in combination with gefitinib, erlotinib, afatinib and dacomitinib is being investigated in cell cultures, subcutaneous & orthotopic xenograft models and clinical tumor samples. Clearly, the use of oncogene-addicted, highly drug-sensitive cell line models to identify escape mechanisms should be a strategy for identifying and overcoming resistance. However, it is of paramount importance that whenever possible, a biopsy is taken at time of cancer recurrence in patients who become resistant to targeted therapies so that these mechanisms can be confirmed or refuted. By identifying how a patient’s cancer becomes refractory to targeted therapies, we will be well positioned to design rational treatment strategies to re-induce remissions. Only with expanding knowledge of these escape mechanisms, availability of drugs targeting escape pathways,and examples of such molecular data informing treatment decisions resulting in good clinical outcome, can re-biopsy and molecular pro fi ling of recurrent drug-resistant tumors be widely incorporated into clinical practice.
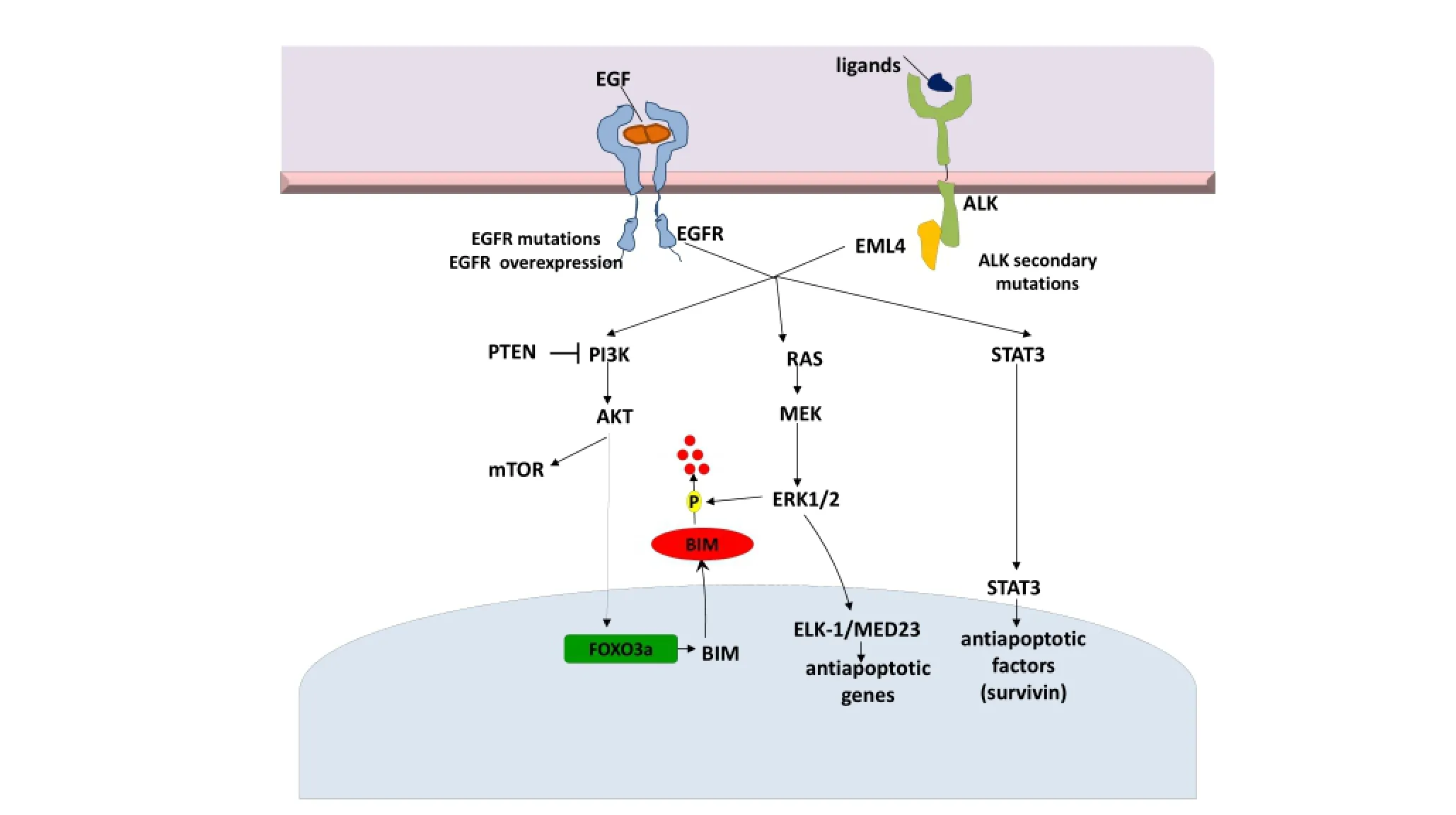
Figure 2 EML4-ALK signaling pathways and potential mechanisms of resistance to targeted therapies. EML4-ALK, echinoderm microtubuleassociated protein-like 4- anaplastic lymphoma kinase.
Acknowledgements
Con fl ict of interest statement
No potential con fl icts of interest are disclosed.
1. Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet 2013;382:720-731.
2. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as fi rst-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239-246.
3. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identi fi cation of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007;448:561-566.
4. Sasaki T, Rodig SJ, Chirieac LR, J?nne PA.e biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010;46:1773-1780.
5. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010;363:1693-1703.
6. Crino L, Kim D, Riely GJ, Janne PA, Blackhall FH, Camidge DR, et al. Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): pro fi le 1005. ASCO Meet Abstr 2011;29:7514.
7. Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol 2012;13:1011-1019.
8. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med 2009;361:958-967.
11. Ou SH. Second-generation irreversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs): a beer mousetrap? A review of the clinical evidence. Crit Rev Oncol Hematol 2012;83:407-421.
12. Ninomiya T, Takigawa N, Ichihara E, Ochi N, Murakami T, Honda Y, et al. Afatinib prolongs survival compared with ge fi tinib in an epidermal growth factor receptor-driven lung cancer model. Mol Cancerer 2013;12:589-597.
13. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013;31:3327-3334.
14. Yang JC, Sequist LV, Schuler MH, Mok T, Yamamoto N, O’Byrne KJ, et al. Overall survival (OS) in patients (pts) with advanced non-small cell lung cancer (NSCLC) harboring common (Del19/ L858R) epidermal growth factor receptor mutations (EGFR mut): Pooled analysis of two large open-label phase III studies (LUXLung 3 [LL3] and LUX-Lung 6 [LL6]) comparing afatinib with chemotherapy (CT). J Clin Oncol 2014;32:abstr 8004.
15. Lee CK, Brown C, Gralla RJ, Hirsh V,ongprasert S, Tsai CM, et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. J Natl Cancer Inst 2013;105:595-605.
16. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26.
17. Spigel DR, Edelman MJ, O’Byrne K, Paz-Ares L, Shames DS, Yu W, et al. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIb or IV NSCLC: Results from the pivotal phase III randomized, multicenter, placebocontrolled METLung (OAM4971g) global trial. J Clin Oncol 2014;32:abstr 8000.
18. Costa C, Molina MA, Drozdowskyj A, Giménez-Capitán A, Bertran-Alamillo J, Karachaliou N, et al.e impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin Cancer Res 2014;20:2001-2010.
19. Faehling M, Eckert R, Kamp T, Kuom S, Griese U, Str?ter J, et al. EGFR-tyrosine kinase inhibitor treatment beyond progression in long-term Caucasian responders to erlotinib in advanced nonsmall cell lung cancer: a case-control study of overall survival. Lung Cancer 2013;80:306-312.
20. Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F, Ha SJ, et al. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancerer2012;11:2254-2264.
21. Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov 2013;3:1404-1415.
22. Berardi R, Santoni M, Morgese F, Ballatore Z, Savini A, Onofri A, et al. Novel small molecule EGFR inhibitors as candidate drugs in non-small cell lung cancer. Onco Targetser 2013;6:563-576.
23. Sequist LV, Soria JC, Gadgeel SM, Wakelee HA, Camidge DR, Varga A, et al. First-in-human evaluation of CO-1686, an irreversible, highly selective tyrosine kinase inhibitor of mutations of EGFR (activating and T790M). J Clin Oncol 2014;32:abstr 8010.
24. Targeting Resistance in Lung Cancer. Cancer Discov 2013;3:OF9.
25. Janne PA, Ramalingam SS, Yang JC, Ahn MJ, Kim DW, Kim SW, et al. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients (pts) with EGFR inhibitor–resistant nonsmall cell lung cancer (NSCLC). J Clin Oncol 2014;32:abstr 8009.
26. Fan W, Tang Z, Yin L, Morrison B, Hafez-Khayyata S, Fu P, et al. MET-independent lung cancer cells evading EGFR kinase inhibitors are therapeutically susceptible to BH3 mimetic agents. Cancer Res 2011;71:4494-4505.
27. Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 2012;18:521-528.
28. Nakagawa T, Takeuchi S, Yamada T, Ebi H, Sano T, Nanjo S, et al. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res 2013;73:2428-2434.
29. Dasari VR, Velpula KK, Alapati K, Gujrati M, Tsung AJ. Cord blood stem cells inhibit epidermal growth factor receptor translocation to mitochondria in glioblastoma. PloS one 2012;7:e31884.
30. Yu B, Liu W, Yu WM, Loh ML, Alter S, Guvench O, et al. Targeting protein tyrosine phosphatase SHP2 for the treatment of PTPN11-associated malignancies. Mol Cancerer 2013;12:1738-1748.
31. You M, Yu DH, Feng GS. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol 1999;19:2416-2424.
32. Sordella R, Bell DW, Haber DA, Seleman J. Ge fi tinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004;305:1163-1167.
33. Lazzara MJ, Lane K, Chan R, Jasper PJ, Ya ff e MB, Sorger PK, et al. Impaired SHP2-mediated extracellular signal-regulated kinase activation contributes to ge fi tinib sensitivity of lung cancer cells with epidermal growth factor receptor-activating mutations. Cancer Res 2010;70:3843-3850.
34. Li L, Han R, Xiao H, Lin C, Wang Y, Liu H, et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res 2014;20:2714-2726.
35. Alvarez JV, Greulich H, Sellers WR, Meyerson M, Frank DA. Signal transducer and activator of transcription 3 is required for the oncogenic e ff ects of non-small-cell lung cancer-associated mutations of the epidermal growth factor receptor. Cancer Res 2006;66:3162-3168.
36. Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest 2007;117:3846-3856.
37. Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A 2010;107:15535-15540.
38. Principe DR, Doll JA, Bauer J, Jung B, Munshi HG, Bartholin L, et al. TGF-beta: duality of function between tumor prevention and carcinogenesis. J Nat Cancer Institute 2014;106:djt369.
39. Dusso A, Arcidiacono MV, Yang J, Tokumoto M. Vitamin D inhibition of TACE and prevention of renal osteodystrophy and cardiovascular mortality. J Steroid Biochem Mol Biol 2010;121:193-198.
40. Li W, Kang Y. A new Lnc in metastasis: long noncoding RNA mediates the prometastatic functions of TGF-β. Cancer Cell 2014;25:557-559.
41. Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, et al. A long noncoding RNA activated by TGF-β promotes the invasionmetastasis cascade in hepatocellular carcinoma. Cancer Cell 2014;25:666-681.
42. Miura K, Wakayama Y, Tanino M, Orba Y, Sawa H, Hatakeyama M, et al. Involvement of EphA2-mediated tyrosine phosphorylation of Shp2 in Shp2-regulated activation of extracellular signal-regulated kinase. Oncogene 2013;32:5292-5301.
43. Maroun CR, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M.e tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol Cell Biol 2000;20:8513-8525.
44. Gusenbauer S, Vlaicu P, Ullrich A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene 2013;32:3846-3856.
45. Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer 2013;13:772-787.
46. Puig de la Bellacasa R, Karachaliou N, Estrada-Tejedor R, Teixidó J, Costa C, Borrell JI. ALK and ROS1 as a joint target for thetreatment of lung cancer: a review. Transl Lung Cancer Res 2013;2:72-86.
47. Barreca A, Lasorsa E, Riera L, Machiorlai R, Piva R, Ponzoni M, et al. Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol 2011;47:R11-R23.
48. Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al.e ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov 2014;4:662-673.
49. Kelly LM, Barila G, Liu P, Evdokimova VN, Trivedi S, Panebianco F, et al. Identi fi cation of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A 2014;111:4233-4238.
50. Tuononen K, Sarhadi VK, Wirtanen A, R?nty M, Salmenkivi K, Knuuila A, et al. Targeted resequencing reveals ALK fusions in non-small cell lung carcinomas detected by FISH, immunohistochemistry, and real-time RT-PCR: a comparison of four methods. Biomed Res Int 2013;2013:757490.
51. Doebele RC. A nice problem to have: when ALK inhibitor therapy works beer than expected. Jorac Oncol 2014;9:433-435.
52. Shaw AT, Kim DW, Mehra R, Tan DS, Felip E, Chow LQ, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med 2014;370:1189-1197.
53. Information ZTcP. East Hanover, New Jersey, USA: Novartis Pharmaceuticals Corporation. April 2014.
54. Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov 2013;3:430-443.
55. Richards MW, Law EW, Rennalls LP, Busacca S, O’Regan L, Fry AM, et al. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical β-propeller domain. Proc Natl Acad Sci U S A 2014;111:5195-5200.
56. Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res 2012;18:6219-6226.
57. Yang JJ, Zhang XC, Su J, Xu CR, Zhou Q, Tian HX, et al. Lung cancers with concomitant EGFR mutations and ALK rearrangements: diverse responses to EGFR-TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res 2014;20:1383-1392.
58. Koyama-Nasu R, Haruta R, Nasu-Nishimura Y, Taniue K, Katou Y, Shirahige K, et al.e pleiotrophin-ALK axis is required for tumorigenicity of glioblastoma stem cells. Oncogene 2014;33:2236-2244.
59. Takezawa K, Okamoto I, Nishio K, J?nne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res 2011;17:2140-2148.
60. Tanizaki J, Okamoto I, Takezawa K, Sakai K, Azuma K, Kuwata K, et al. Combined e ff ect of ALK and MEK inhibitors in EML4-ALK-positive non-small-cell lung cancer cells. Br J Cancer 2012;106:763-767.
Cite this article as:Karachaliou N, Rosell R. Systemic treatment in EGFRALK NSCLC patients: second line therapy and beyond. Cancer Biol Med 2014;11:173-181. doi: 10.7497/j.issn.2095-3941.2014.03.003
Rafael Rosell
E-mail: rrosell@iconcologia.net
Received August 21, 2014; accepted September 6, 2014. Available at www.cancerbiomed.org
Copyright ? 2014 by Cancer Biology & Medicine
 Cancer Biology & Medicine2014年3期
Cancer Biology & Medicine2014年3期
- Cancer Biology & Medicine的其它文章
- Breast metastasis from lung cancer: a report of two cases and literature review
- The gene expression patterns of BMPR2, EP300, TGFβ2, and TNFAIP3 in B-Lymphoma cells
- Neoadjuvant chemoradiotherapy for resectable esophageal cancer: an in-depth study of randomized controlled trials and literature review
- Hepatitis B virus X protein accelerates the development of hepatoma
- Heterogeneity and renal mass biopsy: a review of its role and reliability
- Restricting carbohydrates to fi ght head and neck cancer—is this realistic?