
Room-temperature degradation of o-xylene in simulated air using an online-regenerable plasma-catalysis reactor with low amounts of nanosized noble metals on Co3O4
2023-11-16 05:37:50ShuchengDI狄書成JiachengXU徐家成ShuiliangYAO姚水良JingLI李晶ZuliangWU吳祖良ErhaoGAO高爾豪JialiZHU朱佳麗LianxinDAI戴連欣WeihuaLIU劉衛(wèi)華BuheZHANG張補(bǔ)河andJunweiZHANG張峻維
Plasma Science and Technology 2023年10期
Shucheng DI (狄書成), Jiacheng XU (徐家成), Shuiliang YAO (姚水良),2,?,Jing LI (李晶),2,?, Zuliang WU (吳祖良),2, Erhao GAO (高爾豪),2,Jiali ZHU (朱佳麗),2, Lianxin DAI (戴連欣), Weihua LIU (劉衛(wèi)華),Buhe ZHANG (張補(bǔ)河) and Junwei ZHANG (張峻維)
1 School of Environmental Science and Engineering,Changzhou University,Changzhou 213164,People’s Republic of China
2 Key Laboratory of Advanced Plasma Catalysis Engineering for China Petrochemical Industry,Changzhou 213164, People’s Republic of China
3 Jiangxi Xintai Functional Materials Technology Co.Ltd., Ji’an 343100, People’s Republic of China
Abstract
Keywords: nanosized Pt, Co3O4, o-xylene degradation, operando plasma DRIFTS, Co3+/Co2+ratio
1.Introduction
Volatile organic compounds(VOCs)in the atmosphere cause damage to the environment and human health [1-3].Aromatic hydrocarbons are the dominant category of VOCs in several megacities, accounting for 20%-50% of total VOC emissions[4].The elimination of aromatic hydrocarbons is of great significance from the perspective of atmospheric environment and human health.O-xylene, as a typical aromatic hydrocarbon, has toxic, irritating, teratogenic, and carcinogenic effects [5, 6].Efforts have been devoted to developing o-xylene treatment technologies, including activated carbon adsorption, membrane separation, biological treatment, thermal decomposition, catalytic oxidation, and nonthermal plasma (NTP) [7, 8].Among them, NTP technology has been widely studied and used to control the emission of low-concentration VOCs due to its better removal ability at atmospheric pressure,simple equipment system,low heat loss, and high pollutant tolerance [9, 10].However, the oxidation process of VOCs using NTP technology generates harmful by-products [11, 12].VOC oxidation using NTP combined with catalysts can effectively reduce the generation of harmful by-products, but there are still problems, such as low energy efficiency and low CO2selectivity [13, 14].Recently, transition metal oxides, such as Co3O4, Mn3O4,CeO2, and NiO, have been widely studied due to their low costs and excellent performance in eco-friendly catalytic oxidation of VOCs [15-18].Because of its excellent reducibility, rich electrophilic O species (Oads, O?, or O2?) and oxygen vacancies [19-22], and low formation enthalpy,Co3O4has been widely studied as a catalyst for VOCs degradation [23-25].
Noble metal-supported catalysts have received increasing attention due to their relatively high catalytic performance[26-28].Ren et al reported the catalytic combustion of soot on a series of hydrotalcite-derived CoAlO-supported Ag catalysts and found that metallic silver can highly disperse on the surface of the support due to the interaction between Co and Ag species via electron transfer [29].Qu et al used the hard template method to prepare Pd or Pt catalysts supported on metal oxides with hollow structures for toluene oxidation[30, 31].Ma et al reported the efficient catalytic oxidation of ethylene by gold nanoparticles supported on mesoporous Co3O4at 0°C [32].Ma et al prepared silver supported on a Co3O4catalyst using a simple one-pot solvent method, and found that a Ag/Co3O4catalyst has excellent low-temperature reduction performance due to its more surface-reactive oxygen species and lattice defects[33].Li et al described that the excellent performance of a Pt/Co3O4catalyst was attributed to the interaction between Pt and Co3O4,the presence of Pt in the low valence state, low temperature reduction, and abundant surface reactive oxygen species [34].Wu et al concluded that, although Au/Co3O4has a lower surface area and larger gold grain size, its catalytic activity for the oxidation of toluene and paraxylene is much higher than that of Au/Al2O3and Au/MgO [35].Wang et al showed that the addition of Pd can improve the catalytic activity of mesoporous and bulk Co3O4[36].
In terms of plasma catalysis, Shou et al reported that o-xylene conversion can be greatly increased using a MnO2/Al2O3catalyst even at a low energy density (ED) of 18 J l?1[37].The authors prepared a series of Fe-doped LaMnO3catalysts to control the production of by-products such as O3, N2O, and CO during NTP degradation of VOCs[37].Guillerm et al found a synergistic effect between plasma catalysis and photocatalysis on the removal of gas-phase ammonia (NH3) and hydrogen sulfide (H2S) [38].Aymen et al found that the removal rate of isovaleric acid and trimethylamine could be improved by increasing the energy density or relative humidity level [39].Zhu et al designed a MnOx/γ-Al2O3catalyst mixed with a small amount of Pt nanoparticles, which could decompose 90% o-xylene with 90% CO2selectivity at an ED of 211 J l?1and a reaction temperature of 150°C [40].In 2020, Zhu et al reported that toluene removal performance can be remarkedly improved using a plasma system coupled with Au nanocatalysts [41].The Au catalyst can improve not only VOCs removal performance but also reduce the emission of nanosized by-product particles [42].Significant VOCs removal can be achieved by using a discharge reactor with a small amount of noble metal catalysts [43].These research results give us the inspiration that noble metal catalysts can compensate for the shortcomings of plasma removal efficiency.Furthermore,Zhu et al developed a ‘storage-oxidation’ process to effectively remove formaldehyde using a thermal catalytic oxidation reactor[44].Their findings hint that a duration of regeneration can be incorporated into a plasma system with low COxselectivity, which will improve the catalyst’s performance after regeneration.
In this study,nanosized noble metals(Pt,Pd,and Au)on Co3O4catalysts were used to catalyze the plasma degradation of o-xylene in simulated air.The influences of the loading amounts of noble metals, energy density, and online regeneration on o-xylene degradation were evaluated.The catalysts were characterized using x-ray diffraction (XRD), highresolution transmission electron microscopy(HRTEM),x-ray photoelectron spectroscopy (XPS), a specific surface and porosity analyzer, hydrogen temperature-programmed reduction (H2-TPR), O2temperature programmed desorption(O2-TPD), and operando plasma diffuse reflectance infrared Fourier transform spectroscopy(DRIFTS).The mechanism of plasma catalytic degradation of o-xylene on Pt/Co3O4was proposed.
2.Experiments
2.1.Experimental system
Figure 1(a) shows the experimental system for evaluating plasma catalytic degradation of o-xylene in simulated air.Figure 1(b) shows the dielectric barrier discharge (DBD)reactor with heat insulation using silica wool for the DBD reactor regeneration.The experimental system and the DBD reactor with heat insulation are described in detail in the supplementary material.
2.2.Catalyst preparation
Pt/Co3O4catalysts were prepared using an impregnation method using Co3O4powder(purity 99.9%,Macklin,China)as the support and H2PtCl6(purity 99.995%,Aladdin,China)as the precursor of Pt.The pH of the precursor solution was adjusted to 7 with urea (purity 99%, Shanghai Test, China).The impregnated solutions were macerated overnight at room temperature.The macerated powder was dried at 150°C for 12 h and calcined in room air at 500°C for 3 h.The amounts of Pt loaded on the Co3O4were 0.01 wt%,0.1 wt%,and 0.5 wt% (the mass weight ratio of Pt to Co3O4), denoted as 0.01Pt/Co3O4, 0.1Pt/Co3O4,and 0.5Pt/Co3O4, respectively.PdCl2(Pd content 59.9%,Macklin, China) and HAuCl4(Au content 49.9%, Macklin,China) were used for preparing the Pd/Co3O4and Au/Co3O4,respectively, using the same impregnation method as the Pt/Co3O4catalysts.The prepared catalysts were denoted as 0.01 Pd/Co3O4, 0.1 Pd/Co3O4, 0.5 Pd/Co3O4, 0.01Au/Co3O4,0.1Au/Co3O4, and 0.5Au/Co3O4.

Figure 1.(a) Schematic of the experimental process and (b) DBD reactor with heat insulation for catalyst regeneration.
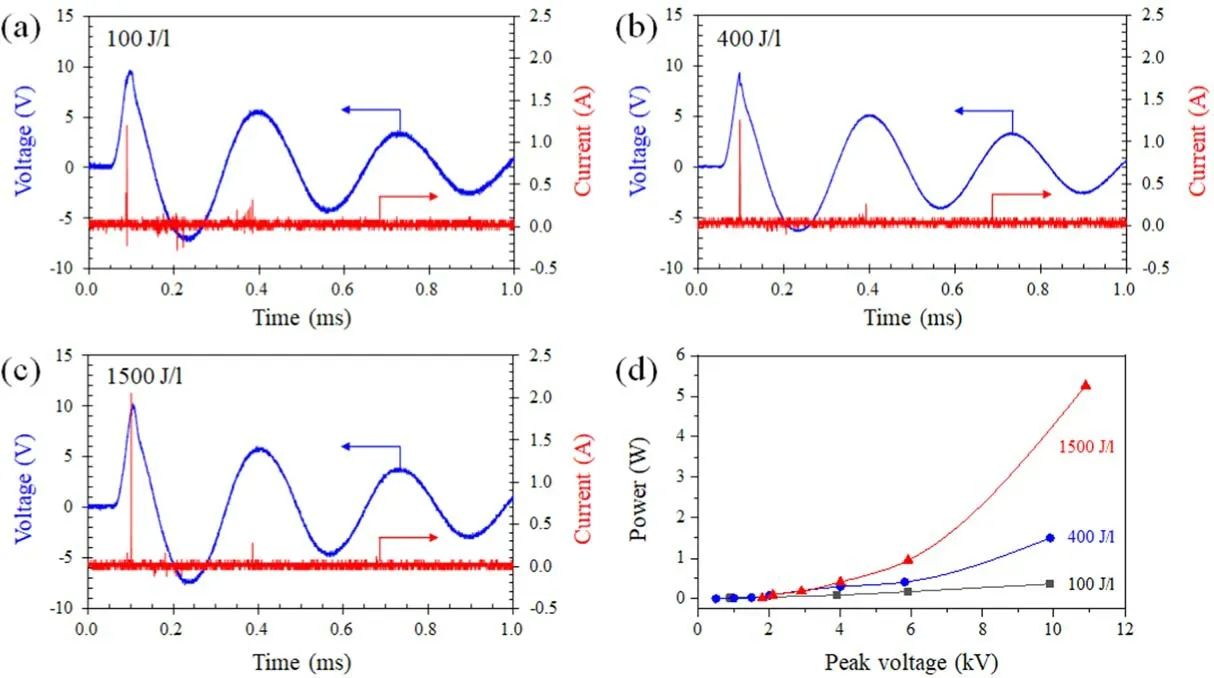
Figure 2.(a)-(c)Typical waveforms of discharge voltage and current using the DBD reactor loaded with the 0.1Pt/Co3O4 catalyst at different voltage pulses.(d) Relation of discharge power and peak voltage.
2.3.Catalytic characterization and calculations
The catalysts were characterized using XRD, HRTEM, a specific surface and porosity analyzer, XPS, H2-TPR,O2-TPD, and operando plasma DRIFTS.Characterization information and calculations are given in detail in the supplementary material.
3.Results and discussion
3.1.Discharge waveforms
Figures 2(a)-(c) show typical waveforms of the discharge voltage and current applied to the DBD reactor filled with the 0.1Pt/Co3O4catalyst; the discharge power was charged by changing the voltage pulses with different peak voltages,where the discharge power was obtained using equation(S1).Figure 2(d) shows the discharge power as a function of peak voltage.When the voltage pulse with a peak voltage of 6.7 kV was applied to the DBD reactor, the current pulse had a maximum value of 0.04 A(figure 2(a)).The peak value of the current pulse increased to 1.64 A by increasing the peak voltage to 10.3 kV;the voltage pulse had a rise time of 28 μs and full width at half maxima of 44 μs(figure 2(e)).When the peak voltage was 9.5 kV, several current pulses in one pulse discharge duration were observed(figure 2(d)),indicating that several micro discharges in the DBD reactor occurred,resulting in more discharge power injected to the discharge space in the DBD reactor.The relation of discharge power and peak voltage (figure 2(f)) revealed that the discharge power did not increase below 8 kV, but increased obviously when the peak voltage was higher than 8 kV, suggesting that 8 kV is the incent voltage for gas discharges using the DBD reactor filled with the 0.1Pt/Co3O4catalyst.
3.2.Catalytic performance
Figure 3(a) illustrates o-xylene conversion using the DBD reactor without a catalyst (none) or with Co3O4or Co3O4loaded with different noble metals (Pt, Pd, and Au) at different EDs.When the DBD reactor was not filled with a catalyst or was filled with Co3O4, o-xylene conversion was only 42% and 60%, respectively, at 100 J l?1, indicating that Co3O4can promote o-xylene conversion.When the DBD reactor was filled with Co3O4loaded with different noble metals, o-xylene conversion significantly increased.At 100 J l?1, o-xylene conversion using the DBD reactor filled with Co3O4loaded with Au or Pd catalysts was up to 95%.O-xylene conversions using the DBD reactor with the 0.1Pt/Co3O4and 0.5Pt/Co3O4catalysts were 98.9% and 99.8%, respectively.These facts imply that noble metals on Co3O4can further promote o-xylene conversion, and the Ptsupported Co3O4catalyst is the best for o-xylene conversion.
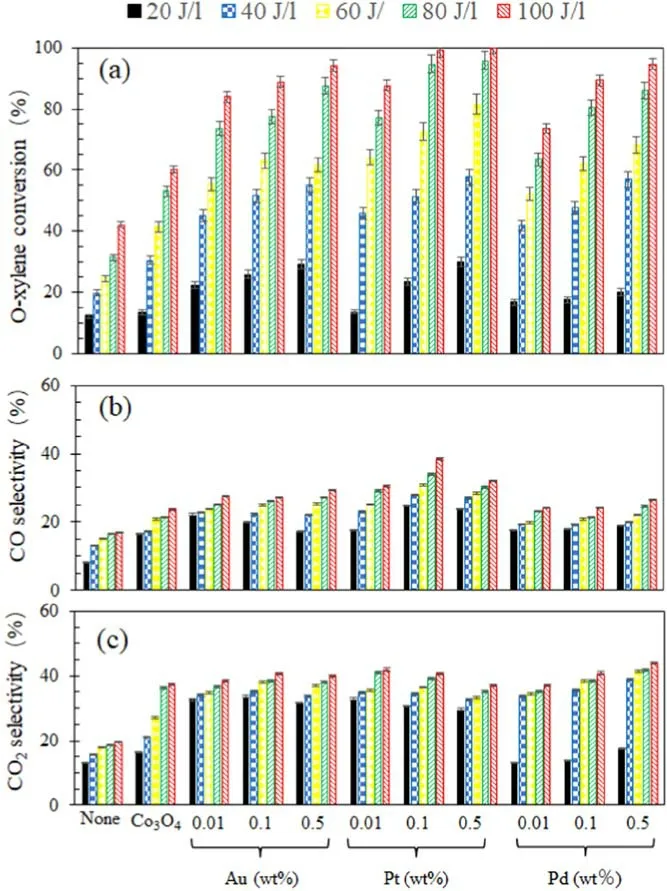
Figure 3.O-xylene conversion (a) CO (b) and CO2 (c) selectivity using DBD reactors without a catalyst (none) and with Co3O4 or noble metal-loaded Co3O4 at various EDs.
Figures 3(b) and (c) show CO selectivity and CO2selectivity using the DBD reactor without a catalyst(none)or with Co3O4or Co3O4loaded with different noble metals (Pt,Pd, and Au) at different EDs.The COxselectivity of the 0.1Pt/Co3O4catalyst was the highest(80%).The selectivities of CO and CO2were 39% and 41%, respectively.
Figure 4(a) shows the effects of heat insulation on COxconcentrations at various elapsed times, where o-xylene(1300 mg m?3)was fed during all elapsed time durations.No obvious difference in CO2concentration was found during the conventional elapsed times of 0-170 min and 280-310 min in which the ED was kept constant (100 J l?1).During the regeneration elapsed time of 170-280 min, the ED was controlled to 1500 J l?1by increasing the pulse frequency; COxconcentrations rapidly decreased to 6940 mg m?3using the DBD reactor with heat insulation and 6500 mg m?3using the DBD reactor without heat insulation.These COxconcentrations were higher than that(about 6200 mg m?3)at 100 J l?1,indicating that degradation of by-products on the catalyst surface had occurred.COxconcentrations using the DBD reactor with heat insulation decreased to constant levels with a time period shorter than that using the DBD reactor without heat insulation,implying that by-product oxidation is faster at a higher temperature since the temperature of the DBD reactor with heat insulation is higher than that without heat insulation(figure 4(d)).
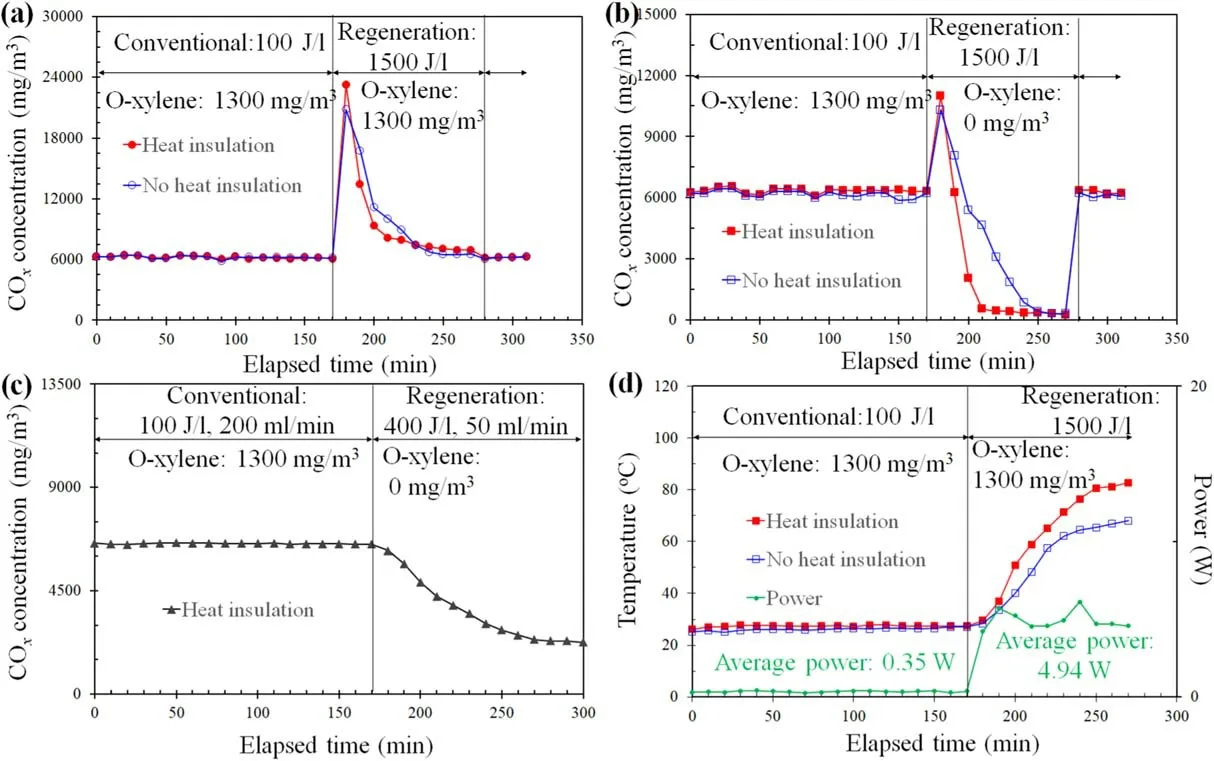
Figure 4.Effect of heat insulation on o-xylene degradation.(a) COx concentrations at 100 J l?1 (conventional degradation) and 1500 J l?1(regeneration), where 1300 mg m?3 o-xylene was continuously fed.(b) COx concentrations at 100 J l?1 (conventional) and 1500 J l?1(regeneration), where the initial o-xylene concentration was 1300 mg m?3 during conventional degradation and 0 mg m?3 during regeneration.(c)COx concentration at 100 J l?1,where the initial o-xylene concentration was 1300 mg m?3 during conventional degradation(0-170 min) and 0 mg m?3 during regeneration (170-300 min).(d) Temperatures and discharge powers during conventional o-xylene degradation and regeneration durations.The total gas flow rate was fixed at 200 ml min?1 except that of 50 ml min?1 during regeneration as given.
Figure 4(b) illustrates the effects of heat insulation on COxconcentrations at various elapsed times, where o-xylene(1300 mg m?3) was not fed during the regeneration elapsed time of 170-280 min at 1500 J l?1.During the regeneration elapsed time,COxconcentrations using the DBD reactor with or without heat insulation decreased to 0 mg m?3, suggesting that the by-products on the catalyst surface could be completely oxidized.
Figure 4(d) presents the temperatures measured at the outside surface point of the quartz tube 5 mm downstream from the discharge zone.The temperature of the DBD reactor with heat insulation was about 1°C-2°C higher than that of the DBD reactor without heat insulation at 100 J l?1; this is due to the electric energy injection from the power supply to the DBD reactor.The temperature of the DBD reactor with heat insulation was 82°C at 310 min elapsed time, 14°C higher than that of 68°C using the DBD reactor without heat insulation.This suggested that more electric energy (average power 4.94 W)at 1500 J l?1was injected to the DBD reactor than that at 100 J l?1, resulting in greater gas temperature increase.
The catalyst regeneration method can also be realized using two alternately operated DBD reactors;when one DBD reactor is used to degrade o-xylene in an exhaust gas, the other is used to regenerate the DBD reactor if the catalyst activity is not satisfied.Figure 4(c) shows conventional o-xylene (1300 mg m?3) degradation at 100 J l?1and a 200 ml min?1gas flow rate for 170 min, followed by catalyst regeneration duration operated at 400 J l?1and 50 ml min?1gas (without o-xylene) for 130 min.COxselectivity was around 6540 mg m?3during conventional degradation and this decreased to 2310 mg m?3at a time of 300 min.This indicates that there are still intermediate products that are not completely oxidized on the catalyst surface.
Figure 5 shows a 14 h long-term o-xylene degradation on a 0.1Pt/Co3O4catalyst at 100 J l?1using the DBD reactor with heat insulation without regeneration or with three regeneration intervals.The regeneration was carried out at a 50 ml min?1gas flow rate with 0 mg m?3o-xylene and 400 J l?1for 90 min,or 1500 J l?1for 30 min.When using the DBD reactor without regeneration, o-xylene conversion and COxselectivity were, respectively, 94% and 81% at an elapsed time of 0 min,which decreased to 83%and 69%at an elapsed time of 840 min.These decreases are due to by-product accumulation on the catalyst surface, resulting in reactivity decay of the catalyst.Figures 5(c) and (d) show that,when using the DBD reactor with regeneration at 400 J l?1for 90 min, o-xylene conversion and COxselectivity decreased from 97% to 91% and from 81% to 74%, respectively.This shows that the DBD reactor with regeneration at 400 J l?1for 90 min can improve o-xylene degradation performance.Figures 5(a) and (b) show that, when using the DBD reactor with regeneration at 1500 J l?1for 30 min, o-xylene conversion and COxselectivity were kept around 95% and 82%,respectively.This fact clearly explains that regeneration effectively maintains stable o-xylene degradation.
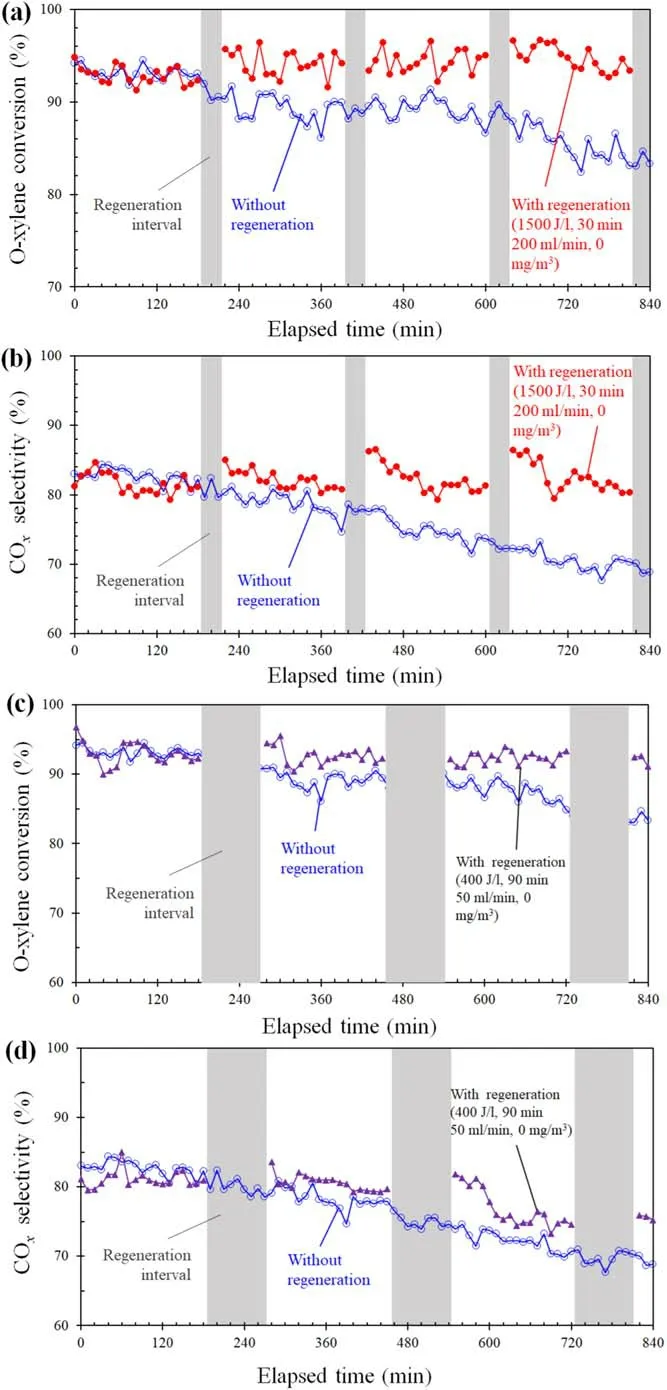
Figure 5.O-xylene conversion((a),(c))and COx selectivity((b),(d))without regeneration or with generation at various elapsed times.Convectional o-xylene degradation was operated at 100 J l?1,200 ml min?1, 1300 mg m?3, and 25°C.Regeneration conditions are listed in the figure.
3.3.By-products on catalyst surfaces
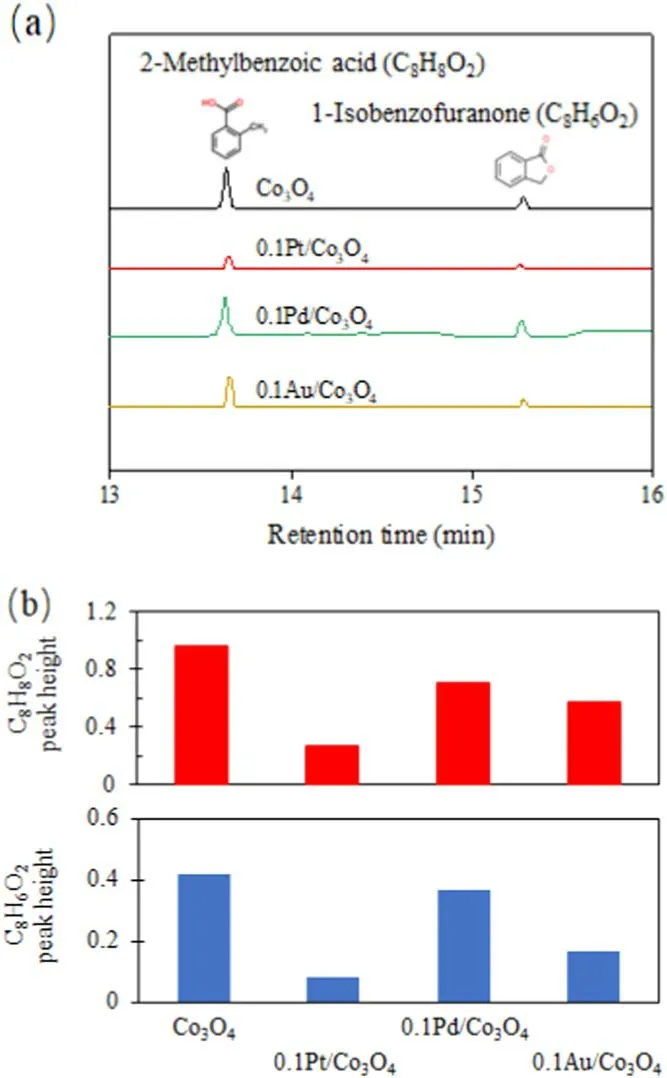
Figure 6.(a) GC-MS spectra of ethanol liquids from Co3O4, 0.1Pt/Co3O4, 0.1 Pd/Co3O4, and 0.1Au/Co3O4.(b) Peak heights of o-methyl benzoic acid(C8H8O2)and isobenzofuranone(C8H6O2)on different catalysts.
Four catalysts were used for o-xylene degradation for 2 h under conditions of 100 J l?1, 25°C, 200 ml min?1gas mixture of 1300 mg m?3, and 20 vol% O2with N2balance.The catalysts were then washed with 20 ml ethanol.The ethanol washing solutions were analyzed using chromatography-mass spectrometry (GC-MS).O-methyl benzoic acid (C8H8O2) and isobenzofuranone (C8H6O2) were found as by-products of all four catalysts(figure 6(a)).Those by-products are obvious from the degradation products of methyl groups in o-xylene molecules.Peak heights of C8H8O2and C8H6O2on 0.1Pt/Co3O4are significantly lower than those on Co3O4(figure 6(b)),indicating that the 0.1Pt/Co3O4catalyst can effectively promote the complete oxidation of o-xylene.
3.4.Energy efficiency
The energy efficiency of o-xylene degradation using plasma catalytic oxidation is an important index of catalytic performance.Figure 7 summarizes the energy efficiencies obtained in this study and compares them with toluene and benzene degradation reported elsewhere.At a low concentration of 1300 mg m?3,the energy efficiency of o-xylene degradation using the 0.1Pt/Co3O4catalyst is within 64.2-116.8 g kW?1h?1, which is close to the current top levels of benzene and toluene degradation reported by Zhu et al [43] and Zhu et al [44].
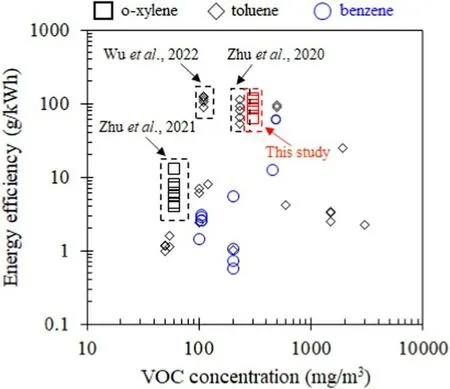
Figure 7.Energy efficiencies for plasma catalytic degradation of aromatic hydrocarbons (o-xylene, toluene, and benzene).

Figure 8.HRTEM images of (a) 0.1Pt/Co3O4, (b) 0.1 Pd/Co3O4,and (c) 0.1Au/Co3O4 catalysts.
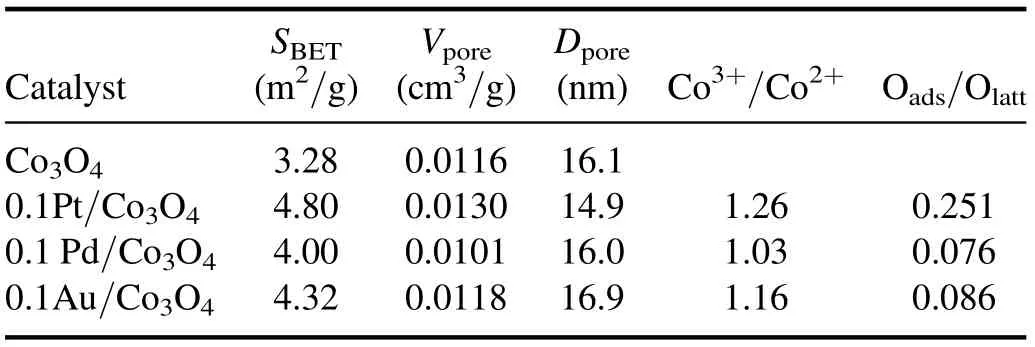
Table 1.BET and XPS data of the catalysts.
3.5.Catalyst characterization
Figure S1 shows the XRD patterns of the Co3O4,0.1Pt/Co3O4,0.1 Pd/Co3O4,and 0.1Au/Co3O4catalysts.All catalysts exhibit high-resolution characteristic diffraction peaks in the range of 2θ from 20° to 80°.The peak positions are 19.0° (111), 31.3° (220), 36.8° (311), 38.6° (222), 44.8°(400), 55.7° (422), 59.3° (511), and 65.3° (440), indicating that the four catalysts all belong to Co3O4(JCPDS card No.42-1476).Due to the good dispersion of low amounts of Pt,Pd, and Au, no diffraction peaks related with Pt, Pd, and Au could be found.Compared with the Co3O4without loading of a noble metal,the intensities of all diffraction peaks were not obviously changed after the loading of Pt,Pd,and Au,but the peak positions of the 0.1Pt/Co3O4, 0.1 Pd/Co3O4, and 0.1Au/Co3O4catalysts shifted by a small angle (-0.1°),implying that the noble metal has a large atomic radius, and part of the noble metal cations were incorporated to the Co3O4lattice [34].
The morphology and microstructure of the 0.1Pt/Co3O4,0.1 Pd/Co3O4, and 0.1Au/Co3O4catalysts are shown in figure 8.The lattice spacing of 0.469-0.479 nm belongs to the(111)crystal plane of standard Co3O4crystal[45].The lattice spacing is about 0.229, 0.231, and 0.237 nm, corresponding to the lattice planes of Pt(111), Pd(111), and Au(111),respectively[46].The size of the Pt particles is about 2.5 nm,which is smaller than that of Pd particles (2.9 nm) and Au particles (3.6 nm).
The N2adsorption-desorption isotherms of the Co3O4,0.1Pt/Co3O4, 0.1 Pd/Co3O4, and 0.1Au/Co3O4catalysts are shown in figure S2.All catalysts exhibit a typical type IV isotherm.The specific surface area (SBET) based on the Brunauer-Emmett-Teller (BET) method, pore size (Dpore), and pore volume(Vpore)are listed in table 1.Co3O4,0.1Pt/Co3O4,0.1 Pd/Co3O4, and 0.1Au/Co3O4have pore sizes of 16.1,14.9, 16.0, and 16.9 nm, and specific surface areas of 3.28,4.80, 4.00, and 4.32 m2g?1, respectively.
Figure 9(a) shows the XPS spectra of the Co 2p of the 0.1Pt/Co3O4, 0.1 Pd/Co3O4, and 0.1Au/Co3O4catalysts.The Co 2P3/2spectrum has two components at 779.7 and 781.2 eV, and the Co 2P1/2spectrum has two peaks at 794.8 and 796.3 eV, which are assigned to Co3+and Co2+cations,respectively [47].The results confirm the mixed valence of Co species in the catalyst.The spin-orbit splitting energy between Co 2P3/2and Co 2P1/2is 15.1 eV, which indicates that the phase on all catalyst surfaces is Co3O4with Co3+and Co2+[48].The molar ratio of Co3+/Co2+was quantitatively calculated, and the results are shown in table 1 and figure 9(d).The molar ratios of Co3+/Co2+on 0.1Pt/Co3O4,0.1 Pd/Co3O4, and 0.1Au/Co3O4are 1.265, 1.207, and 1.156, respectively.Since Co3+cations are active sites for VOC oxidation, whereas Co2+cations are almost inactive[49], the 0.1Pt/Co3O4catalyst with the highest Co3+/Co2+molar ratio has the most active sites on the catalyst surface for o-xylene degradation.
Figure 9(b)shows the XPS spectra of O 1 s,which prove the presence of surface oxygen species.The peaks at 529.8,531.5, and 532.1 eV correspond to lattice oxygen (Olatt),surface adsorbed oxygen (Oads), and surface adsorbed water or OH groups [15], respectively.Electrophilic Oadsspecies plays an important role in VOC catalytic oxidation due to its better mobility on the catalyst’s surface than lattice oxygen[45].Table 1 shows that the molar ratios of Oads/Olatton the catalyst surfaces have an order of 0.1Pt/Co3O4>0.1Au/Co3O4> 0.1 Pd/Co3O4.The 0.1Pt/Co3O4catalyst has the highest Oads/Olattmolar ratio, which ensures the best performance of o-xylene degradation.
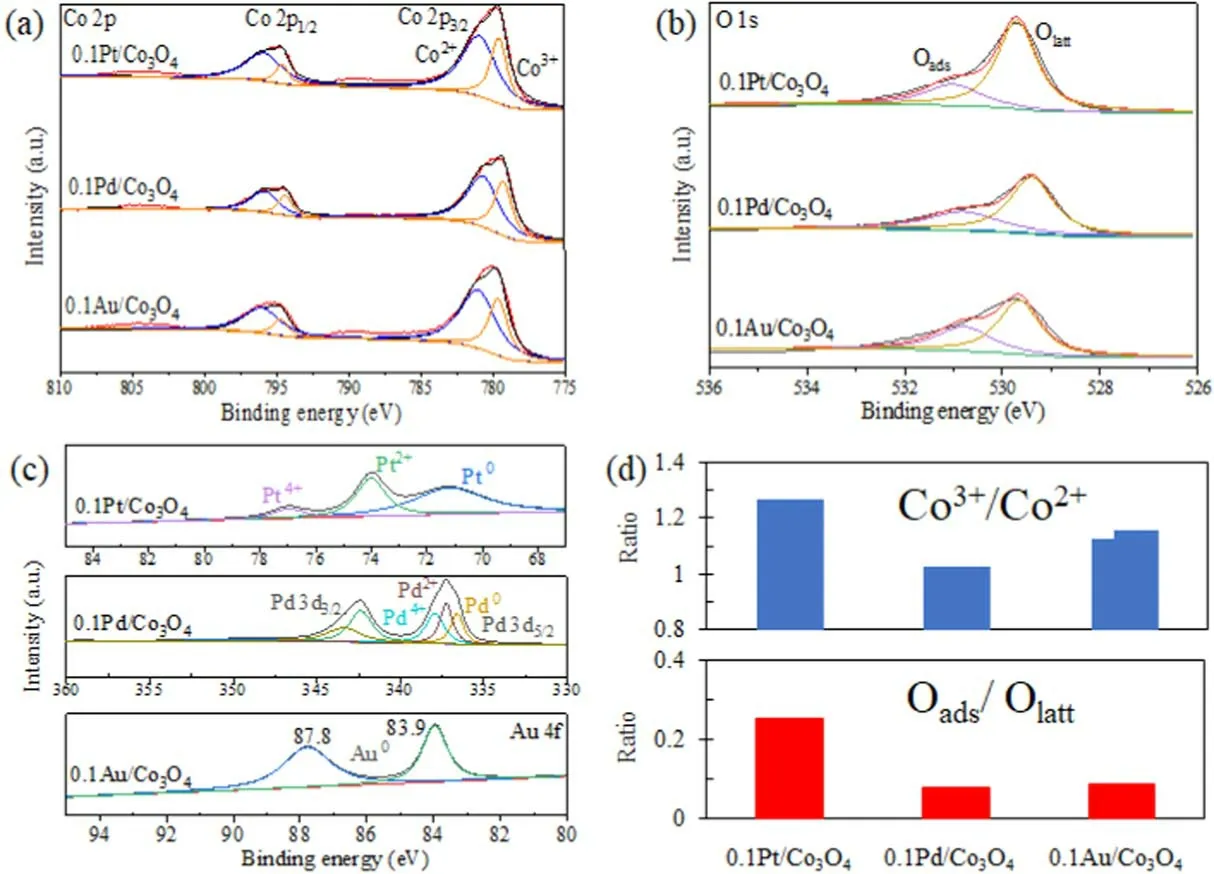
Figure 9.(a) Co 2p, (b) O 1 s, (c) Pt 4 f, Pd 3d, and Au 4 f XPS spectra of different catalysts.(d) Ratios of Co3+/Co2+ and Oads/Olatt.
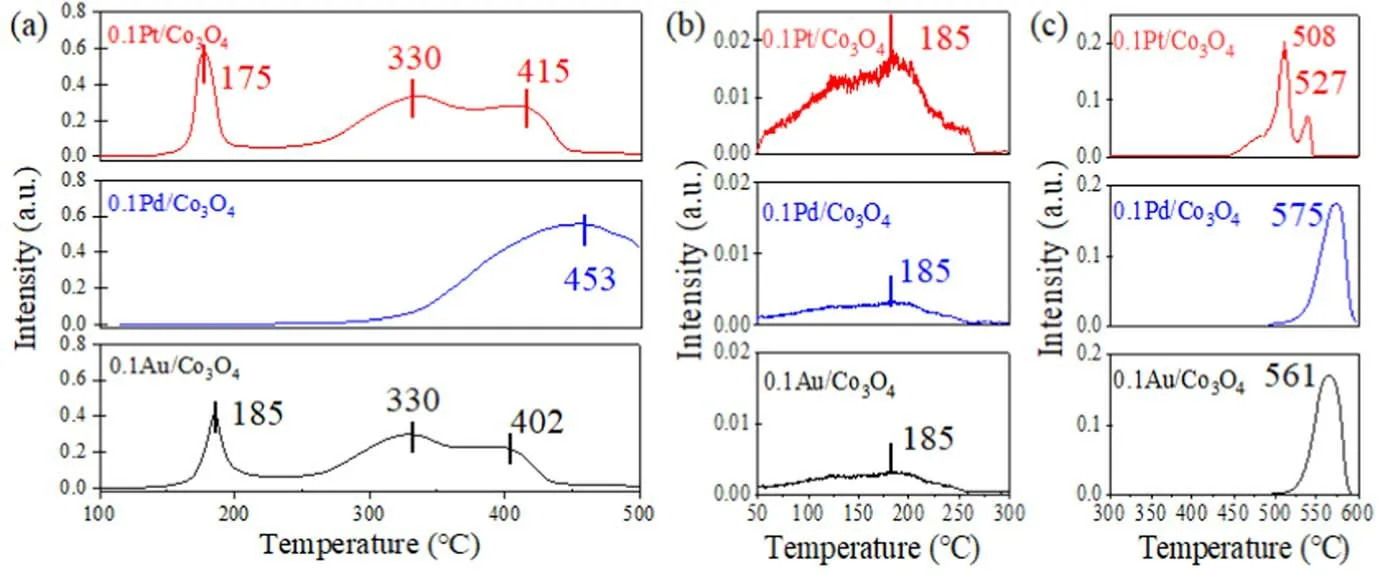
Figure 10.(a) H2-TPR profiles.(b) and (c) O2-TPD profiles.
The Pt4f spectrum of the 0.1Pt/Co3O4catalyst is divided into three component peaks (figure 9(c)), which are located near 71.1, 74.1, and 77.0 eV, respectively, corresponding to Pt0,Pt2+,and Pt4+species[50].In the 0.1 Pd/Co3O4catalyst,Pd can also exist in Pd0, Pd2+, and Pd4+with binding energies of 335.1-335.9, 336.8-337.2, and 337.8-338.3 eV,respectively [51].Peaks corresponding to metal Au0(about 83.9 and 87.8 eV) were observed in the Au 4 f spectra of the 0.1Au/Co3O4catalyst.In all catalysts, Pt0, Pd0, and Au0substances can promote oxygen activation [52].
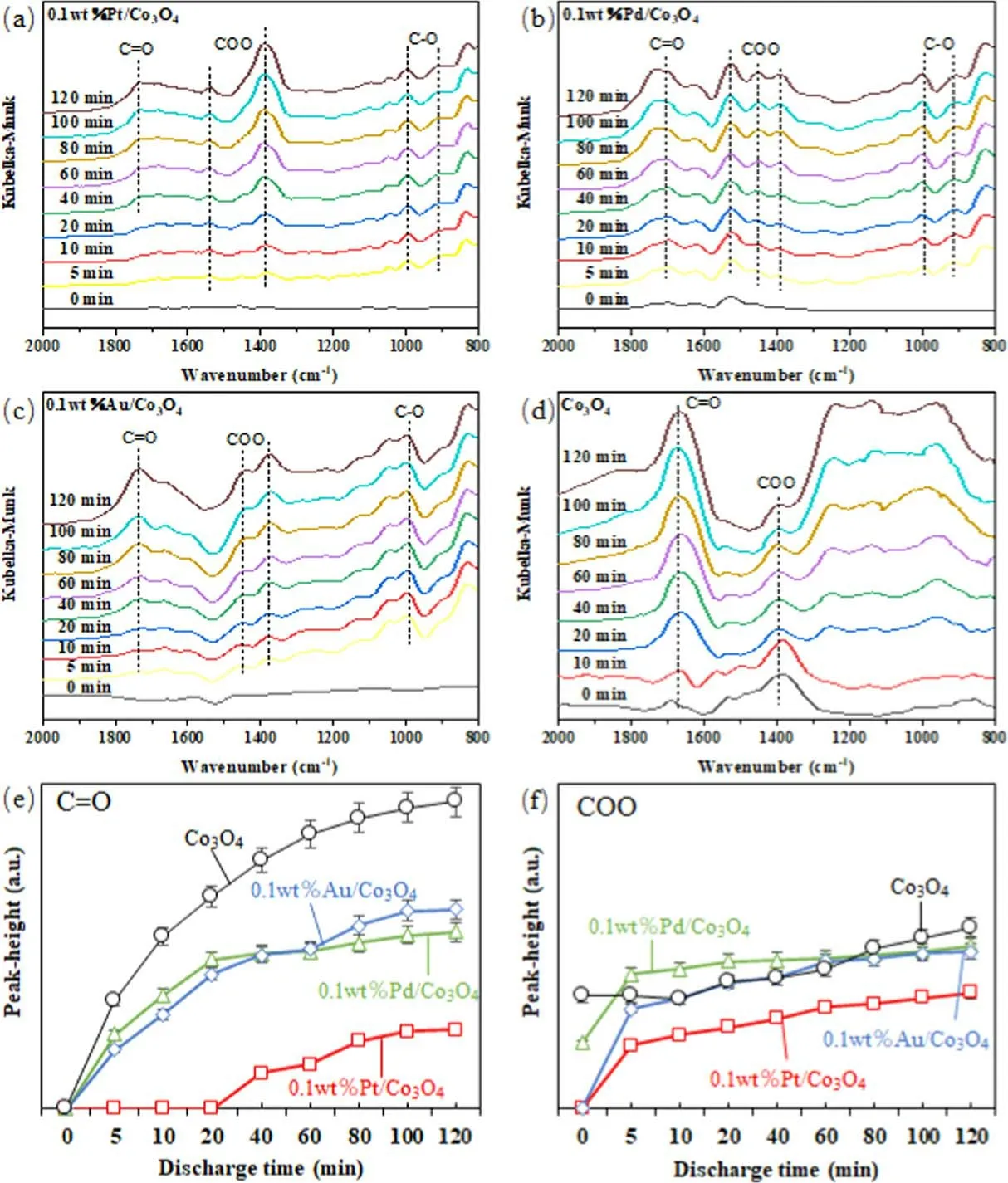
Figure 11.DRIFTS spectra during o-xylene degradation over (a) 0.1Pt/Co3O4, (b) 0.1 Pd/Co3O4, (c) 0.1Au/Co3O4, and (d) Co3O4; peak heights of C=O and COO on different catalysts ((e), (f)).
The above results show that the 0.1Pt/Co3O4catalyst not only has high molar ratios of Co3+/Co2+and Oads/Olatt, but also the presence of Pt0makes the surface reactions of Oadsand Olatta great contribution to o-xylene degradation.
Figure 10(a) shows the H2-TPR profiles of the 0.1Pt/Co3O4, 0.1 Pd/Co3O4, and 0.1Au/Co3O4catalysts.The 0.1 Pd/Co3O4catalyst has only one high temperature reduction peak at 453°C, indicating that its reduction reactivity at a low temperature is poor.This can be proved by the fact that the by-products on 0.1 Pd/Co3O4have higher peak heights than those on 0.1Pt/Co3O4and 0.1Au/Co3O4.The 0.1Pt/Co3O4and 0.1Au/Co3O4catalysts have three distinct reduction peaks (at 175°C, 185°C, and 330°C), which are due to the attribution of Co3+reduction to Co2+and Co2+reduction to metal Co and the reduction of interfacial oxygen species [34].The 0.1Pt/Co3O4catalyst has a peak at the lowest temperature of 175°C, suggesting that 0.1Pt/Co3O4has the highest low temperature activity toward H2oxidation.The 0.1Pt/Co3O4catalyst has reduction peaks in the temperature range of (250-450) °C, and the temperature range is shifted toward the low temperature range in comparison with that of 0.1 Pd/Co3O4due to the interaction between Pt and Co3O4.
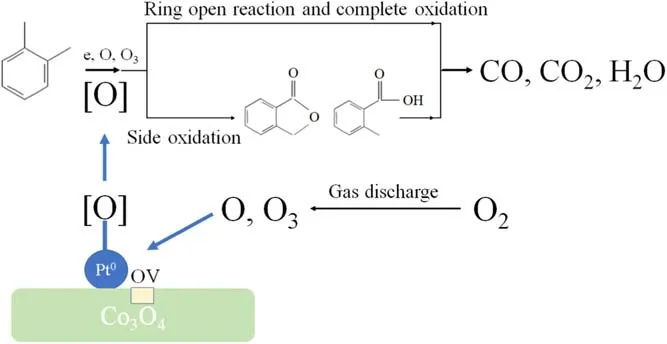
Figure 12.The mechanism of Pt/Co3O4 promotion on o-xylene degradation.
O2-TPD profiles can explore surface chemisorbed oxygen at a temperature lower than 250°C,surface lattice oxygen in a temperature range of (250-600) °C, and bulk lattice oxygen at a temperature higher than 600°C[53].As shown in figures 10(b) and (c), the desorption peaks of surface chemisorbed oxygen at 185°C can be found from the 0.1Pt/Co3O4, 0.1 Pd/Co3O4, and 0.1Au/Co3O4catalysts,where the peak height from 0.1Pt/Co3O4is the highest,proving that the 0.1Pt/Co3O4catalyst has the most surface chemisorbed oxygen [28].
DRIFTS spectra during o-xylene degradation on the 0.1Pt/Co3O4, 0.1 Pd/Co3O4, 0.1Au/Co3O4, and Co3O4catalysts at various discharge times are shown in figures 11(a)-(d), respectively.The peaks at 1780, 1420, 1390, and 980 cm-1were observed after 5 min.The peak at 1780 cm?1is due to the ν(C=O) stretching vibration of aromatic acids[54,55].The peaks at 1420 and 1390 cm?1correspond to the asymmetric carboxylic acid (COO) group and symmetric ν(COO)stretching vibrations[56,57].The peak at 980 cm?1is assigned as the ν(C-O) stretching vibration of alkoxide species [58].The above peaks indicate that the formation of o-methyl benzoic acid (C8H8O2) and isobenzofuranone(C8H6O2) found from GC-MS analysis is reasonable.
The peak heights of C=O and COO groups on different catalysts are illustrated in figures 11(c) and (f).The peak heights of the C=O group on the 0.1 Pd/Co3O4,0.1Au/Co3O4, and Co3O4catalysts gradually increase with increasing discharge time, while that on the 0.1Pt/Co3O4catalyst increases after 20 min discharge time.At a fixed discharge time,the peak heights of the C=O and COO groups on the 0.1Pt/Co3O4catalyst are obviously lower than those on the 0.1 Pd/Co3O4, 0.1Au/Co3O4, and Co3O4catalysts.This fact indicates that 0.1Pt/Co3O4can efficiently promote the oxidation of by-products on the catalyst surface to CO2.
3.6.Mechanism of o-xylene degradation
The mechanism of o-xylene degradation in the DBD reactor over 0.1Pt/Co3O4is proposed in figure 12.O2in the gas mixture is first decomposed to O atoms by impact with energized electrons in the gas discharge space,where O atoms can react with O2to form O3.O and O3can be adsorbed on the 0.1Pt/Co3O4surface and converted to surface reactive oxygen species [O].The nanosized Pt has interfaces with Co3O4and maintains a high Co3+/Co2+ratio at these interfaces,which benefits the formation of oxygen vacancies(OV)on the Co3O4surface.Energized electrons(e),O,O3,and[O]contribute to o-xylene degradation possibly via two paths:one is the ring open reaction (to CO, CO2, and H2O) and the other is side oxidation (to by-products).The by-products of o-methyl benzoic acid (C8H8O2) and isobenzofuranone(C8H6O2) can be further oxidized to CO, CO2, and H2O.From figure S3, it can be seen that the ozone concentrations from the Pd/Co3O4and Au/Co3O4catalysts at a fixed ED were lower than that from Co3O4, suggesting that the inhibition of ozone formation and ozone decomposition occurred on the Pd/Co3O4and Au/Co3O4catalysts, which benefitted complete o-xylene oxidation.This is consistent with the results of GC-MS [59-61].
4.Conclusions
The catalytic degradation of o-xylene in simulated air using a DBD reactor with 0.1Pt/Co3O4, 0.1 Pd/Co3O4, 0.1Au/Co3O4, and Co3O4was evaluated at room temperature.The catalysts were characterized using XRD, XPS, BET,HRTEM, H2-TPR, O2-TPD, and operando plasma DRIFTS.The main conclusions are as follows.
(1) Pt,Pd,and Au on Co3O4can obviously enhance o-xylene degradation using a DBD reactor at room temperature.
(2) The 0.1Pt/Co3O4catalyst has the highest o-xylene degradation performance, and o-xylene conversion and COxselectivity were 98.9% and 76.5%, respectively, at 100 J l?1.This energy efficiency is in the top level in comparison to those reported in the literature.
(3) O-xylene degradation performance can be improved by incorporating regeneration durations.A stable o-xylene degradation performance was obtained by regenerating the catalyst online using a heat-insulated reactor operated at 1500 J l?1.
(4) A Co3O4and Pt synergistic interaction in the 0.1Pt/Co3O4catalyst induces more oxygen vacancies to form more Oads, a high Co3+/Co2+ratio, and Pt0content, all of which improve complete o-xylene oxidation.
(5) Operando plasma DRIFTS results clearly demonstrated the complete o-xylene oxidation and proved that Pt played a key role.
Acknowledgments
This study was supported by National Natural Science Foundation of China (No.12075037) and Research and Application Service Platform Project of API Manufacturing Environmental Protection and Safety Technology in China(No.2020-0107-3-1).
猜你喜歡
Acta Mathematica Scientia(English Series)(2022年4期)2022-08-25 08:52:14
美術(shù)界(2022年4期)2022-04-26 11:07:00
中學(xué)生博覽(2021年11期)2021-07-15 22:45:22
中學(xué)生博覽(2021年13期)2021-07-14 01:24:29
Acta Mathematica Scientia(English Series)(2021年3期)2021-06-17 13:59:16
中學(xué)生博覽(2021年3期)2021-03-08 02:25:51
中學(xué)生博覽(2021年1期)2021-03-04 19:49:14
黃河·黃土·黃種人(華夏文明)(2019年10期)2019-11-01 07:31:30
今日農(nóng)業(yè)(2019年10期)2019-01-04 04:28:15
中學(xué)課程輔導(dǎo)·教學(xué)研究(2017年11期)2017-09-23 12:22:39
 Plasma Science and Technology2023年10期
Plasma Science and Technology2023年10期
- Plasma Science and Technology的其它文章
- 3D fluid model analysis on the generation of negative hydrogen ions for negative ion source of NBI
- Enhancing surface adhesion of polytetrafluoroethylene induced by two-step in-situ treatment with radiofrequency capacitively coupled Ar/Ar+CH4+NH3 plasma
- Etching characteristics and surface modification of InGaSnO thin films under Cl2/Ar plasma
- Pulsed gas-liquid discharge plasma catalytic degradation of bisphenol A over graphene/CdS: process parameters optimization and O3 activation mechanism analysis
- A homogeneous atmospheric pressure air plasma in a 10mm gap based on a threeelectrode configuration
- Effect of gas flow on the nanoparticles transport in dusty acetylene plasmas