
Novel mutations in PDE6A and CDHR1 cause retinitis pigmentosa in Pakistani families
2021-12-17 02:42:18MuhammadDawoodSiyingLinTajUdDinIrfanUllahShahNiamatKhanAbidJanMuhammadMarwanKomalSultanMahaNowshidRaheelTahirAsifNaveedAhmedMuhammadYasinEmmaBapleAndrewCrosbyShamimSaleha
Muhammad Dawood, Siying Lin, Taj Ud Din, Irfan Ullah Shah, Niamat Khan, Abid Jan,Muhammad Marwan, Komal Sultan, Maha Nowshid, Raheel Tahir, Asif Naveed Ahmed,Muhammad Yasin, Emma L. Baple, Andrew H. Crosby, Shamim Saleha
1Department of Biotechnology and Genetic Engineering,Kohat University of Science and Technology (KUST), Kohat 26000, Khyber Pakhtunkhwa, Pakistan
2Medical Research, RILD Wellcome Wolfson Centre (Level 4), Royal Devon and Exeter NHS Foundation Trust, Exeter,Devon EX2 5DW, UK
3Department of Ophthalmology, KMU Institute of Medical Sciences (KIMS) Kohat 26000, Khyber Pakhtunkhwa, Pakistan
Abstract
● KEYWORDS: autosomal recessive retinitis pigmentosa;PDE6A; CDHR1; variants; Pakistani families
INTRODUCTION
Retinitis pigmentosa (RP) refers to a group of inherited retinal disorders, characterized by progressive degeneration of photoreceptors and retinal pigment deposits resulting in vision loss. Initial symptoms include night blindness, decreased visual acuity and progressive loss of peripheral vision. The majority of RP cases (about 70%-80%) are non-syndromic,where the eye is exclusively affected[1]. The most common forms of syndromic RP, where RP occurs in conjunction with extraocular and/or systemic features, include Usher syndrome and Bardet-Biedl syndrome[2]. The age of onset may vary from childhood to adolescence or early adulthood. The estimated frequency of RP is about 1 in 4000 individuals[3], however,autosomal recessive forms of RP (arRP) have been more frequently reported in Israeli, Saudi, south Indian and Pakistani populations with high rates of consanguinity and endogamy[4-6].Approximately 130 genes have been associated with RP,including about 90 genes for non-syndromic RP and about 40 genes for syndromic forms[7](RetNet; https://sph.uth.edu/retnet/). In addition, about 59 genes are known to underlie other forms of inherited retinal dystrophies (RetNet; https://sph.uth.edu/retnet/). Genes commonly reported to cause arRP includeABCA4(MIM #601691),CRB1(MIM #604210),CDHR1(MIM #609502),PDE6A(MIM #180071),PDE6B(MIM #180072),RHO(MIM #180380),RP1(MIM #603937),SPATA7(MIM #609868),EYS(MIM# 612424),RP1L1(MIM #608581),TULP1(MIM #602280), andUSH2A(MIM #608400); among themCRB1,PDE6A,PDE6B,RP1, andTULP1are the most commonly mutated genes in consanguineous/endogamous families of Pakistani origin[6,8-9].
Despite high genetic heterogeneity, variants in known disease genes are identified in only about 60% of reported cases of RP[10]. Identification of novel genes or genetic variants underlying RP in this cohort of genetically unsolved cases is important in facilitating a better understanding of disease progression and for the development of novel therapies, and,to this end, significant progress has been achieved in recent years through adaptation of high-throughput DNA sequencing technologies[11-12].
In this study, we investigated an endogamous family and a consanguineous family with arRP from Khyber Pakhtunkhwa region of Pakistan. Our clinical and genomic investigations including high-throughput DNA sequencing technologies identified two novel candidate disease variants, a frameshift sequence alteration inPDE6A, as well as splice site sequence alteration inCDHR1that segregated with the disease phenotype in the affected families.
SUBJECTS AND METHODS
Ethical Approval The study was approved by the Ethical Committee of Kohat University of Science and Technology(KUST; Pakistan), and the study was carried out in accordance with the Declaration of Helsinki. Informed written consent was obtained for participation in the study from families’ members and parents of the minor children.
Subjects Family 1 extending over two generations with four affected and six unaffected members, was recruited from the Khyber Pakhtunkhwa region of Pakistan. Further,Family 2 extending over four generations and comprising of eight living affected and 34 unaffected members, was also recruited from same region of Pakistan (Figures 1A and 2A).In Family 1 parents of affected individuals were from same tribe (endogamous family) and in Family 2 parents of affected individuals were cousins (consanguineous family). Blood samples were collected from recruited affected and unaffected individuals, and all affected individuals were clinically evaluated by local ophthalmologists for obtaining medical and family histories and clinical assessment. The clinical assessment procedures used were funduscopic examination and visual acuity measurements performed using Snellen charts,as well as colour fundus photography in selected affected individuals, for diagnosis of RP.
Genetic Analysis Genomic DNA from blood samples was extracted using the ReliaPrep? kit (Blood gDNA Miniprep System, Promega) according to the manufacturer’s protocol. To identify the causative disease variant, whole exome sequencing was performed on a single affected individual in each family(subject II:4 in Family 1 and II:10 in Family 2 in Figures 1 and 2 respectively) to develop a profile of rare sequence variants present at low frequency in publicly available population databases. Coding regions were captured by HiSeq2000 using paired-end (2×100) protocol at a mean coverage depth of 30×at the Otogenetics Corporation (Norcross, GA, USA). The Agilent SureSelect Human All ExonV4 (51 Mb) enrichment kit was used for exome enrichment. The sequence reads were aligned to the human genome reference sequence [hg19] and read alignment, variant calling, and annotation were performed by DNAnexus (DNAnexus Inc., Mountain View, CA; https://dnanexus.com).
Allele-specific primers were designed using Primer3 web software (PDE6AF: 5-AGAGATCCACACTTGCCATCA-3,PDE6AR:5-GAGCGCAAACACCCAGATTT-3;CDHR1F:5-GGCACTCACAGTCCATTCAC-3,CDHR1R:5-CAAGTTGAATTTGGATGG-3) to evaluate segregation of variantsviadideoxy sequencing. Polymerase chain reaction(PCR) was undertaken for all recruited family members using allele-specific primers following standard conditions,with products sequenced by Source BioScience LifeSciences(https://www.sourcebioscience.com/). Pathogenicity of the identified sequence variation in theCDHR1was analyzed using CRYP-SKIP (https://cryp-skip.img.cas.cz/), Human Splicing Finder (https://www.genomnis.com/access-hsf), and Transcript Inferred Pathogenicity Score (TraP) (http://trapscore.org/) specialized prediction software. The effect of thePDE6Agene variant on protein structure was predicted using Phyre2 software (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index). To compare and correlate thePDE6AandCDHR1gene variants with phenotype, all reported variants were retrieved from HGMD (http://www.hgmd.cf.ac.uk/ac/search.php), OMIM (https://www.ncbi.nlm.nih.gov/omim/),ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and PubMed(https://www.ncbi.nlm.nih.gov/pubmed/) databases.
RESULTS
Subjects Pedigree analysis of recruited Pakistani families suggested an autosomal recessive mode of disease inheritance in the affected families (Figures 1 and 2). In Family 1, the four affected individuals II:1, II:2, II:4 and II:8 were aged 20, 19, 16 and 4y at time of first examination, whilst in Family 2, the eight affected individuals II:1, II:7, II:10, II:13,II:15, III:15, III:16, and III:18 were aged 51, 46, 43, 40, 36,30, 18, and 12y respectively. Night blindness and decreased visual acuity were prominent common clinical features in all affected members of both investigated families (Table 1). On the basis of basic clinical ophthalmic assessment, arRP was the major finding in all affected members. Affected member had the classical RP phenotypesi.e.,waxy disc pallor, attenuated vessels and bone spicules in both the fundi. In addition, the affected members of the Family 2 had features of macular atrophy along with RP. There was a wide range of severity of clinical features noted in affected members of both families(Figure 3).
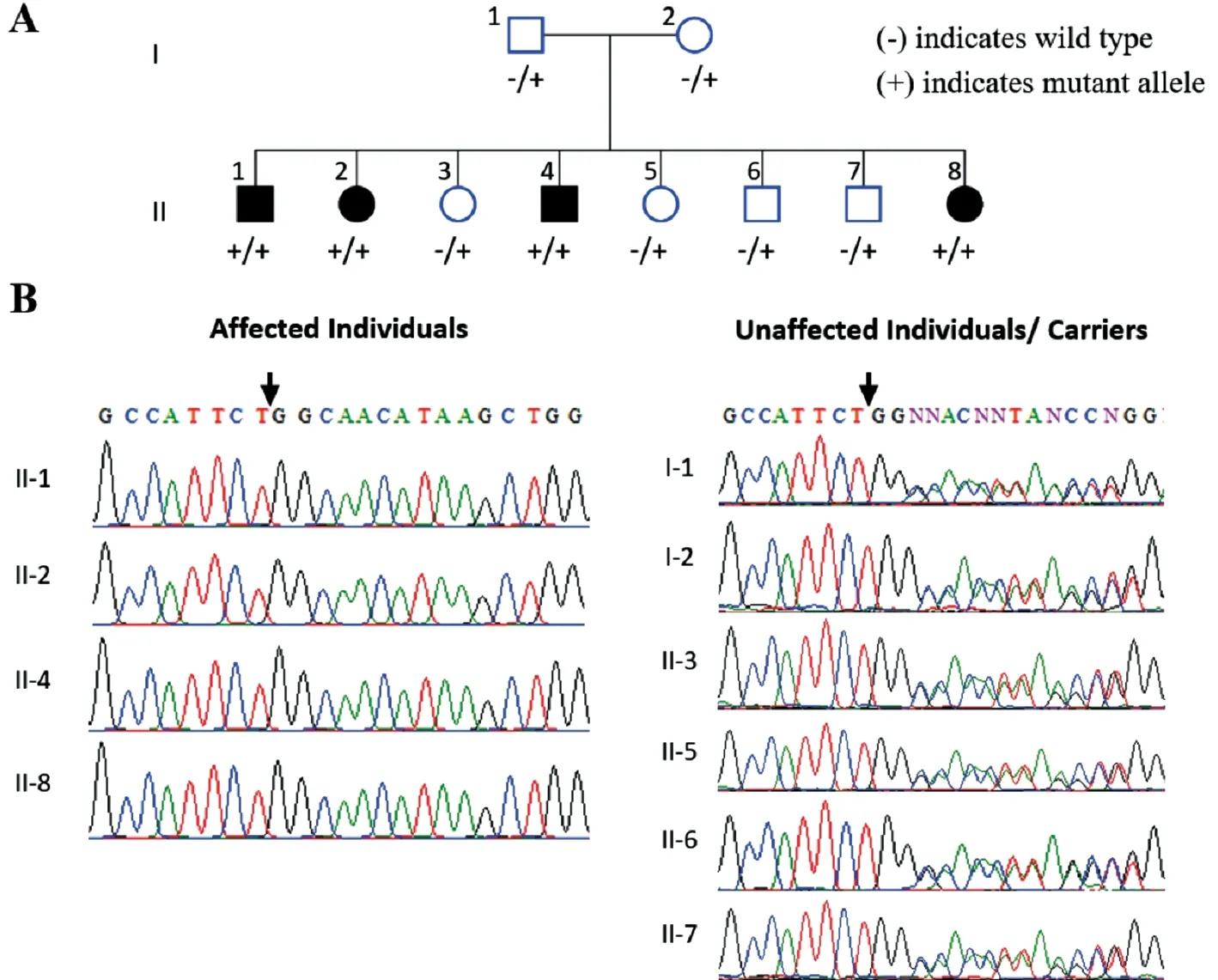
Figure 1 Family 1 pedigree and genetic findings A: Pedigree shows segregation of the PDE6A variant identified. B: Sequence chromatograms show the PDE6A [NM_000440.2:c.1054delG, p.(Gln352Argfs*4); Chr5:g.149286886del (GRCh37)] variant in affected (homozygous) and phenotypically normal (heterozygous carriers) members of investigated family.
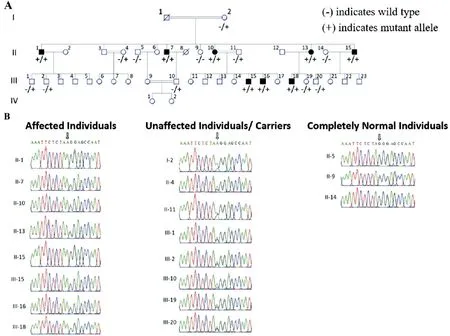
Figure 2 Family 2 pedigree and genetic findings A: Pedigree shows segregation of CDHR1 variant identified. B: Sequence chromatograms show the CDHR1 [NM_033100.3:c.1168-1G>A, p.(?); Chr10:g.85968484G>A (GRCh37)] gene variant in the affected and phenotypically normal members of investigated family.
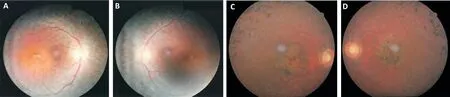
Figure 3 Colour fundus photographs of a single affected individual in each family A, B: Right and left eyes of II:4 in Family 1; C, D: Right and left eyes of II:7 in Family 2. In both affected individuals, typical features of RP are noted, including optic disc pallor, attenuated retinal vessels and mid-peripheral bone spicule pigmentation. Additionally, both affected individuals display features of macular involvement.
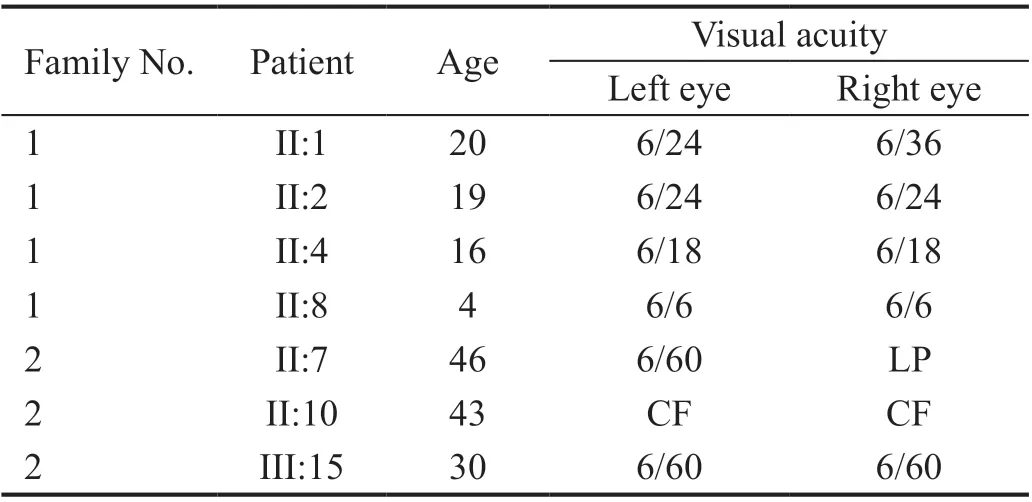
Table 1 The age and visual acuity of the affected individuals of both families
Genetic Findings Initial analysis of exome data excluded previously described variants in genes known to cause ocular disease. Variants were then assessed and filtered for rare, non-synonymous exonic or splice variants, with a population frequency of <0.01 in control databases (including the Genome Aggregation Database; gnomAD, the Exome Aggregation Consortium; ExAC, and the 1000 Genomes Project). A single candidate novel homozygous frameshift variant [NM_000440.2:c.1054delG; Chr5:g.149286886del(GRCh37)] was identified in exon 7 ofPDE6A(Figure 1)in Family 1. This single base pair deletion is predicted to result in a frameshift followed by a premature stop codon(p.Gln352Argfs*4). In Family 2, a novel splice site substitution[NM_033100.3:c.1168-1G>A, Chr10:g.85968484G>A(GRCh37)] was identified at the splice acceptor site in intron 11 of theCDHR1gene (Figure 2). In silico analysis of c.1168-1G>A using Human Splicing Finder, CRYP-SKIP and TraP predicted it to result in activation of a new cryptic splice acceptor site within the exon 12 sequence which may lead to alternative splicing and partial skipping of exon 12 or skipping of entire exon 12 sequence during splicing. Both thePDE6Ap.(Gln352Argfs*4) as well as theCDHR1c.1168-1G>A variants are absent in gnomAD. The sequence variants in families 1 and 2 segregate as expected for an autosomal recessive condition in each family (Figures 1 and 2).
Variants reported to date inPDE6Afor arRP in Pakistani families are summarized in Table 2[8,13-16]. A schematic representation of pathogenic variants identified to date inPDE6AandCDHR1are shown in Figure 4A-4D).
DISCUSSION
Non-syndromic RP cases are frequently reported in South Asian populations, particularly from Pakistan and Southern India, with autosomal recessive modes of inheritance observed in up to 95% of RP cases[6]. The high rates of consanguineous marriages (20% to 50%), as well as endogamous marriages increasing intracommunity genetic homogeneity, both contribute to the high burden of arRP in these populations[6,17-20]. RP is characterized by significant clinical and genetic heterogeneity,leading to diagnostic challenges. Over half of genes currently known to cause non-syndromic RP were identified in South Asian populations[6]. Therefore, the large multigenerational consanguineous/endogamous families of South Asian populations are powerful resources for genetic studies of arRP,facilitating the identification of novel variants or disease genes that contribute to the disease[6].
In current study, genetic analyses identified novel candidate disease variants inPDE6A[c.1054delG, p.(Gln352Argfs*4)]andCDHR1[c.1168-1G>A, p.(?)] that segregated appropriately with autosomal recessive disease phenotype in Pakistani families. Previously, variants inPDE6A(p.Arg102Ser,p.Arg257*, c.1408-2A>G, p.Arg544Trp, p.Tyr700Leufs*21 and c.2028-1G>A) have been described in some Pakistani families with arRP[8,15]. To best of our knowledge, only one mutation (c.1463delG, p.Gly488Alafs*18) in theCDHR1gene for retinal dystrophies have been reported in families of Pakistani origin[21-22]. In contrast, disease causing mutations(p.Pro574Ala, c.1485+2T>C, p.Tyr547*, p.Tyr140*,p.Ala128Gly and p.Asp696Alafs) in theCDHR1gene have been detected to cause retinal dystrophies in consanguineous/endogamous Spanish, Chinese, Saudi, Lebanese and Israeli populations[23-27]. Previous studies have found thatAIPL1(MIM#604393),CRB1(MIM #604210),SEMA4A(MIM #607292),RPGRIP1(MIM # 605446),TULP1(MIM #602280)RP1(MIM #603937),PDE6B(MIM #180072), CNGB1(MIM# 600724) andRHO(MIM #180380) are most commonly mutated genes in Pakistani families to cause arRP and other retinal disorders[6,28]. These studies have also provided evidence of founder effect of few genes’ mutations in Pakistani population. In this context, theAIPL1mutationp.Trp278* was reported previously founder mutation in Pakistani families with juvenile arRP[8,29-32]. In addition,TULP1mutations p.Lys489Arg and p.Thr380Ala were found to be more commonly mutated in Pakistani families with arRP, thus may be referred as Pakistani founder mutations[32-35]. In contrast, no founder effect has been reported so far ofPDE6Amutations identified previously and in current study in Pakistani origin families with arRP[8,15]. However, elucidation of founder effect of reportedPDE6Amutations in our population may have an impact on efficient diagnosis and counselling of affected families.
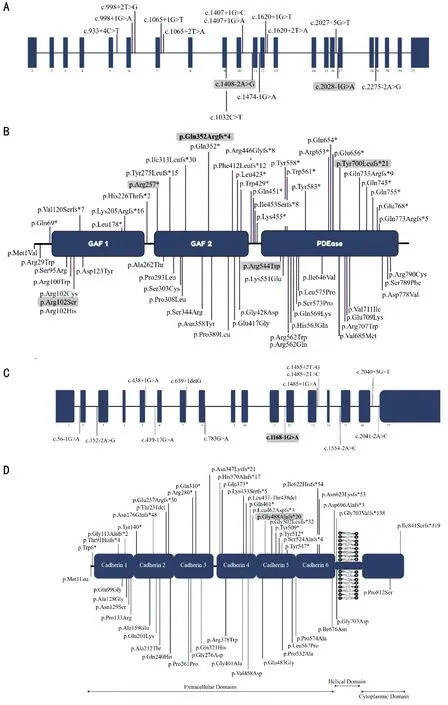
Figure 4 PDE6A and CDHR1 variants associated with arRP A, C: Schematic representation of exons of the PDE6A and CDHR1 gene highlighting the positions of all disease causing mutations identified to date; B, D: Domains of PDE6A and CDHR1 predicted protein[UniProtKB - P16499; UniProtKB - Q96JP9] respectively, highlighting the positions of all disease associated variants identified to date. The variants identified in Pakistani origin patients previously and both the variants identified in this study are in bold and grey color.
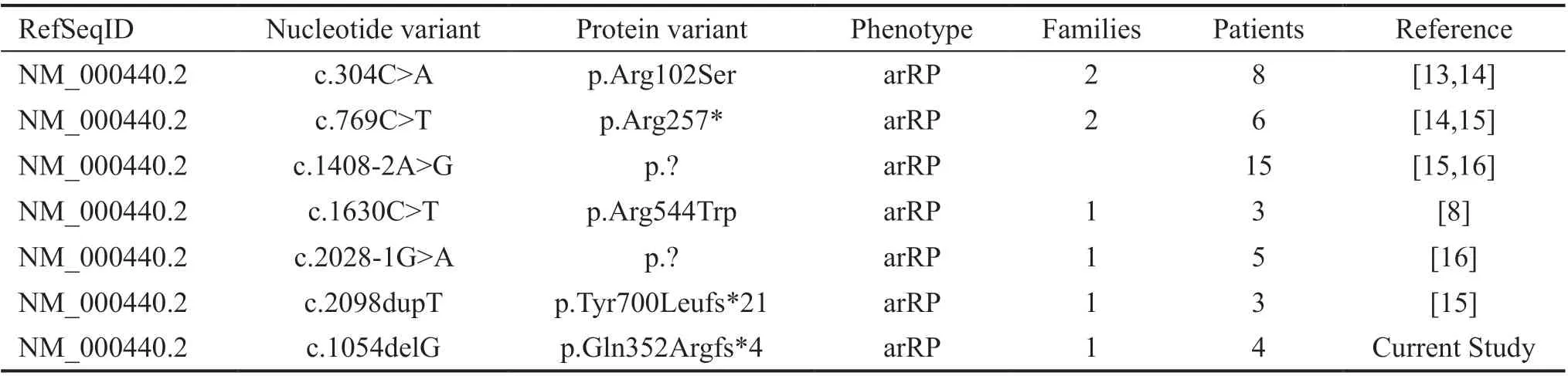
Table 2 Summary of all reported PDE6A variants associated with arRP in Pakistani families
The translated PDE6A protein contains 2 GAF domains(GAF-1; 73-222 amino acids and GAF-2; 254-431 amino acids) and a cyclic nucleotide phosphodiesterase (PDEase)domain (483-816 amino acids; UniProtKB - P16499).Normally, thePDE6Agene expresses the alpha subunit of phosphodiesterase 6 complex (PDE6), the other members of the PDE6 complex are a beta subunit (PDE6B) and two inhibitory gamma subunits (PDE6G). The PDE6 complex is directly involved in visual phototransduction pathway, and is localized on the disc membrane of the rod photoreceptor cells[36-37]. During phototransduction in rod photoreceptors,absorption of a photon of light leads to isomerization of rhodopsin. This isomerized rhodopsin activates transducin(a heterotrimeric G-protein) in the disc membrane, which activates the PDE complex, leading to hydrolysis of cyclic guanine monophosphate (cGMP) molecules into GMP molecules and resulting in a fall in the level of cGMP inside the rod photoreceptors. As a result, the cGMP-gated cation channel in the plasma membrane closes and membrane is hyperpolarized[37]. Loss of function in thePDE6Aprotein leads to impairment of the photo-transduction pathway and an increase in cGMP levels. This increase in cGMP levels results in the death of rod and cone photoreceptors cells, leading to the clinical manifestations of RP[38-40].
The identified variant p.Gln352Argfs*4 in the GAF-2 domain like other nonsense variants, is predicted to cause loss of function ofPDE6Avianonsense-mediated decay[41]. Disease causing variants inPDE6Aorthologues have also been reported to cause RP[42-43]. Loss of function variants inPDE6Aare known to cause arRP. Such as a study conducted by Nairand colleagues reported a novel 2-bp frameshift variant p.Ile452Serfs*7 inPDE6Ain Emirati population[44]. The identified variant (p.Gln352Argfs*4) in current study and the variant p.Arg257* reported previously by Riazuddinet al[15]lie in GAF-2 domain that results in loss of function ofPDE6Ain phototransduction pathway and cause arRP in affected individuals. Previously, Jespersgaard and colleagues found a nonsense variant at same amino acid position (p.Gln352*) inPDE6Athat also caused arRP[45].
TheCDHR1protein is an important member of calciumdependent cadherin superfamily, and is involved in the cell adhesion[46].CDHR1is abundantly expressed in the retina,and variants in this gene have been associated with retinal dystrophies ranging from cone dystrophy to RP[47]. In addition toCDHR1, three other members of the cadherin superfamily,PCDH15,CDH3andCDH23, have been implicated in retinal dystrophies[48-50].CDHR1contains six cadherin domains(Cadherin 1; 36-135 amino acids, Cadherin 2; 136-246 amino acids, Cadherin 3; 247-353 amino acids, Cadherin 4; 359-472 amino acids, Cadherin 5; 473-576 amino acids and Cadherin 6; 573-688 amino acids), one transmembrane domain (701-725 amino acid) and one intracellular domain (773-859 amino acids; UniProtKB-Q96JP9). In retinal cells,CDHR1protein is abundantly localized to the junction of inner and outer region of rod and cone photoreceptors[51-52]and is thought to be crucial for the normal structure and survival of the rod and cone photoreceptors. Loss of function ofCDHR1may affect the structure of rod and cone cells that may be the leading cause of retinal dystrophies.
TheCDHR1splice site variants such as c.1168-1G>C that has been reported in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/variation/955272/) and c.1168-1G>A identified in current study are expected to affect the canonical splice sequence at the intron 11 3’ acceptor splice site, and is predicted to result in skipping of exon 12 ofCDHR1or activation of a cryptic splice site in exon 12 ofCDHR1.Exon 12 codes for 50 amino acids in the cadherin 4 domain ofCDHR1and in case of skipping of this exon may lead to the loss of 50 amino acids inCDHR1protein. Similarly, a previous study also reported disease causing nonsense variant in same cadherin 4 domain of theCDHR1that resulted in a premature truncated protein[53]. Missense variants causing retinal dystrophies have been identified in all 6 cadherin repeat domains ofCDHR1, supporting the importance of these domains in maintaining normalCDHR1protein function and hence structural integrity of the photoreceptor cells.
In summary, the identification of novel variants inPDE6AandCDHR1in the present study consolidates the key role of these genes in the pathogenesis of arRP and contributes to the expanding spectrum of disease-causingPDE6AandCDHR1variants. In addition, our study highlights the fact that diseasecausingPDE6AandCDHR1variants, although rare, can cause arRP in Pakistani endogamous and consanguineous families.
ACKNOWLEDGEMENTS
The authors would like to thank the patients and their family members for participation in this study.
Foundation:Supported by the Kohat University of Science and Technology, Kohat, Pakistan and RILD Wellcome Wolfson Centre (Level 4), Royal Devon and Exeter NHS Foundation Trust, UK.
Conflicts of Interest:Dawood M, None; Lin S, None; Din TU, None; Shah IU, None; Khan N, None; Jan A, None;Marwran M, None; Sultan K, None; Nowshid M, None;Tahir R, None; Ahmed AN, None; Yasin M, None; Baple EL, None; Crosby AH, None; Saleha S, None.
 International Journal of Ophthalmology2021年12期
International Journal of Ophthalmology2021年12期
- International Journal of Ophthalmology的其它文章
- Comparison of trifocal toric and bifocal toric intraocular lens implantation in patients with cataract and high corneal astigmatism
- Comparison of perioperative parameters in one-handed rotational phacoemulsification versus conventional phacoemulsification and femtosecond laser-assisted cataract surgery
- Decreased retinal microvasculature densities in pterygium
- Fourier analysis of corneal Scheimpflug imaging: clinical use in keratoconus
- Establishment of a prediction tool for ocular trauma patients with machine learning algorithm
- Role of orthoptics and scoring system for orbital floor blowout fracture: surgical or conservative treatment