
Structure-based simulations complemented by conventional all-atom simulations to provide new insights into the folding dynamics of human telomeric G-quadruplex*
2021-07-30 07:43:40YunQiangBian邊運(yùn)強(qiáng)FengSong宋峰ZanXiaCao曹贊霞JiaFengYu于家峰andJiHuaWang王吉華
Chinese Physics B 2021年7期
Yun-Qiang Bian(邊運(yùn)強(qiáng)), Feng Song(宋峰), Zan-Xia Cao(曹贊霞),Jia-Feng Yu(于家峰), and Ji-Hua Wang(王吉華)
Shandong Key Laboratory of Biophysics,Institute of Biophysics,Dezhou University,Dezhou 253023,China
Keywords: molecular dynamics simulation,structure-based model,all-atom model,DNA G-quadruplex
1. Introduction
Functions of proteins and nucleic acids are supported by their folded structures. Therefore, correct folding is a crucial step for them to achieve their physiological significance.In recent years, the folding problem of DNA G-quadruplex(GQ) structures,[1-3]a family of noncanonical four-stranded helices built by G-quartets (Fig. 1(a)), has attracted much attention.[4-25]One of the most controversial systems is the human telomeric (Htel) GQ, which plays relevant roles in genome stability and anticancer therapies.[26-28]It is well known that the Htel GQ is extremely polymorphic.[29]Influenced by the kind of monovalent cations and other factors, the Htel sequence can fold into a parallel, antiparallel or hybrid GQ topology (Fig. S1 in the Supplementary Material).[30-42]This outcome indicates that the folding landscape contains several well-separated deep basins,and multiple different folds may coexist at the thermodynamic equilibrium.As a result, Htel GQ folding is expected to follow a typical kinetic partitioning or multiple-pathway model, according to which only a fraction of the molecules fold to the native structure rapidly and the other fractions become trapped in various competing basins of attraction.[43]
For Htel GQ, the identification of competing folds is difficult in experiments, since their measurable signals may mutually overlap. However, several studies have indeed detected the kinetic partitioning folding behavior involving different folds.[44-46]In a study conducted by Marchand and coworkers on the folding of various Htel and c-myc sequences,using electrospray mass spectrometry and CD experiments,they suggested a competition between four three-quartet GQs and two two-quartet GQs.[44]In another study reported by Bessiet al., the sequence Htel24 (dTTGGG[TTAGGG]3A)was straightly demonstrated by real-time NMR spectroscopy to fold via a kinetic partitioning between the hybrid-1 (H1)and hybrid-2(H2)GQs in the presence of K+(Fig.1(b)).[46]Of particular interest is that the H2 conformation was unveiled to be kinetically more accessible. Despite of these explicit experimental demonstrations, understanding of the detailed kinetic partitioning folding process is still largely incomplete in experiments due to the temporal and spatial limits of techniques. Molecular dynamics(MD)simulations are deemed to be powerful methods to provide complementary information for experiments. Accumulated studies have demonstrated that such simulations indeed provide basic insights into the structural dynamics by focusing on specific regions of the conformational space.[47-51]For instance, by conducting extensive atomistic simulations, Stadlbaueret al.have systematically investigated the structural stability and lifetimes of various potential folding intermediates of GQs, such as G-hairpins,[47]G-triplexes[48]and late-stage intermediates.[49]However, because the kinetic partitioning folding landscape is considerably rugged, contemporary all-atom simulations are still not sufficient to sample the full folding landscape. Interestingly,in recent studies, coarse-grained models have been validated to be effective in investigation of Htel GQ folding.[52-54]For example,using a high-resolution coarse-grained DNA model,Cragnoliniet al.provided a direct view of the interconversion between different GQ folds.[53]Another example is the hybrid atomistic structure-based model,a variant of the structurebased model or Gˉo-like model which is widely used to study the folding of proteins and nucleic acids.[55-58]We have validated the effectiveness of the hybrid atomistic structure-based model in the investigation of GQ folding.[59,60]This model is more appropriate to study the folding of DNA GQs than the traditional structure-based model. It is known that the reorientation of glycosidic configurations is an important aspect to complicate the folding of DNA GQs. Traditional structurebased models cannot correctly characterize this conformational transition because local conformations are strongly biased to the native conformations.
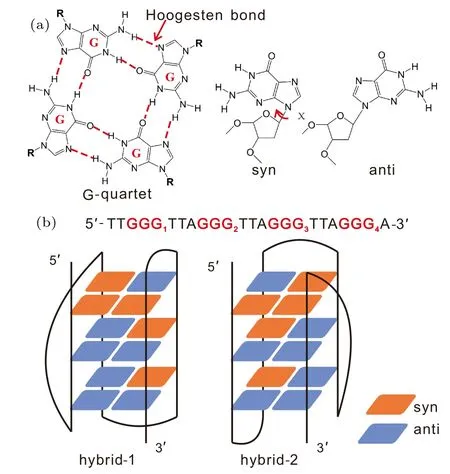
Fig. 1. Schematic illustration for the G-quartet and the two GQ topologies formed by human telomeric sequence Htel24.(a)The structure of a G-quartet stabilized through eight Hoogsten bonds between four guanine bases. Also shown are the syn and anti glycosidic configurations of a guanine base depending on the χ dihedral. (b) The sequence and two GQ folds of Htel24.The red and blue squares denote the guanine nucleotides with syn and anti configurations,respectively.
In contrast,the hybrid atomistic structure-based model removes the favor of local native conformations as it borrows descriptions of local interactions from classical force fields,and thus it is capable of describing the reorientation of glycosidic configurations more accurately. By developing a doublebasin hybrid atomistic structure-based model, we studied the competition between H1 and H2 conformations in the folding of Htel24 and successfully reproduced the experimentally observed kinetic preference of H2.[60]
As mentioned above,exploring the full folding landscape of Htel GQ is still unaffordable for conventional atomistic simulations. Although coarse-grained models can improve the sampling efficiency, they are suffered from the problem of low accuracy in comparison with conventional atomistic models. To alleviate this problem, employing a multiscale framework that merges the high accuracy of all-atom models and the high efficiency of coarse-grained models becomes a reasonable way. Developing such multiscale methods has also been an attractive issue in the field of biophysics. Available examples are the resolution-exchange MD simulations[61,62]and the atomic interaction-based coarse-grained(AICG)model.[63]In the resolution-exchange method,multiple replicas with different levels of coarse-graining are employed and they are allowed to exchange with each other through a metropolis criterion. This strategy is able to largely improve the sampling efficiency of all-atom models because the conformation space sampled by the low-resolution replica can also be visited by the all-atom replica during the simulation. Similar methods include the multigraining MD[65]and the multiscale essential sampling.[66,67]Compared with the strategy of coupling coarse-grained and atomistic models directly in a simulation as in the resolution-exchange method,the AICG model adopts a different strategy that refines the coarse-grained potential function by performing a set of all-atom simulations. For example, one can define new coarse-grained angle and dihedral potentials to characterize the flexibility of protein chains more accurately by extracting structural information from all-atom simulations on corresponding fragments.[64]Several other methods, such as the Bolzmann inversion[68]and the force-match,[69,70]also employ this refine framework. Other multiscale simulation strategies can be found elsewhere.[71-75]In this study,we developed a multiscale simulation framework to couple the hybrid atomistic structure-based model and the all-atom model by two steps. First, a large number of conformations were acquired by performing structure-based simulations. Then,a set of conformations were selected as seeds to conduct large-scale conventional all-atom simulations after reconstructing the structures to atomistic models. It is worth noting that the reconstruction is easy because only hydrogen atoms are not considered in the atomistic structure-based model. Afterwards, the folding thermodynamics and kinetics were analyzed by applying a combination of the Markov state model (MSM)[76]and the transition path theory (TPT).[77]Gromacs-4.6.2[78]was used to conduct all the simulations,and PyEmma[79]was employed to build the MSM and to perform the TPT analysis. For simplicity, four G-repeats in the sequence were labeled as GGG1-GGG4in the 5′-3′direction(Fig.1(b)). Consequently,the substructure established by GGG1and GGG2was simplified to GGG12,and the other substructures were similarly named. The results show that the allatom simulations can complement the findings of the hybrid atomistic structure-based model and can provide more atomistic insights into the folding landscape.
2. Theoretical method
2.1. Hybrid atomistic structure-based model
In this section, we introduce the hybrid atomistic structure-based model briefly. Details have been presented elsewhere.[59,60]All the heavy atoms of the system are retained in this model,and the solvent is implicitly modeled by the Langevin dynamics. Since modeling ions explicitly is still challenging in coarse-grained models,the current model does not consider the roles of ions. The Hamiltonian includes two parts:Vhybrid=Vff+Vsb,(1)
whereVffis the potential of local interactions andVsbis the potential of long-range structure-based interactions.Vffincludes the bonded potential, the angular potential, the proper and improper dihedral potential and the 1-4 short-range potential. In this study,descriptions of local interactions were borrowed from the AMBER DNA force field OL15.[80-82]This force field was chosen because it merges all the newest modifications of DNA dihedral potential which guarantees us to acquire more accurate results. To describe the coexistence of two different folds of Htel24, which are characterized as the H1 and H2 GQ conformations,Vsbmerges the information of these two structures:
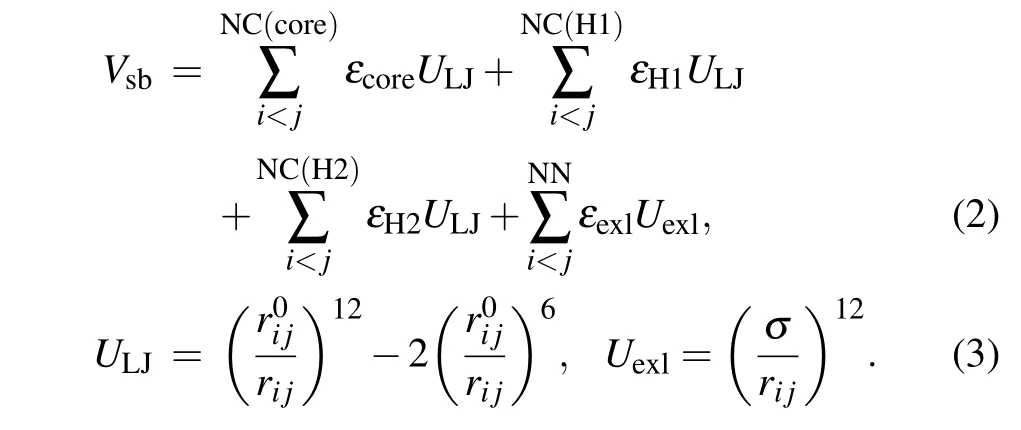
ULJis a 12-6 Lennard-Jones attraction potential for the atom pairs forming native contacts(NC),andUexlis a generic hardcore repulsion potential for the nonnative (NN) atom pairs.It should be noted that the native contacts were defined as those atom pairs within a cutoff of 0.4 nm in the native structure. The native contacts shared by the H1 and H2 conformations were named“core”contacts,while those formed only in one of the two conformations were called specific H1 or H2 contacts. Here,εcore,εH1,εH2,εexl,r0i jandσare the user parameters. Determination of these parameters can be found in our previous study.[60]It is worth noting that various models have been developed to study multi-state biological processes.[83-88]We built the double-basin Hamiltonian by employing a mix-contact map strategy,[83-86,88]which has been validated to be successful in investigation of protein folding and allostery.In this framework,native contacts are classified into different libraries depending on whether they form in both reference structures(core native contacts)or merely form in one reference structure (“specific” native contacts). These information are then integrated into the long-range interaction term with different potential strengths. This is different from the other widely used double-basin model developed by Okazakiet al.[87]In that model, two structure-based singlebasin potentials are built asV(R|R1)andV(R|R2)+ΔV.Ris the coordinates of protein structure andR1(R2)represents the coordinates of the reference structure. ΔVis a parameter modulating the relative stability of the reference structures. Then,a smoothed double-based potentialVMBis obtained by solving a secular equation.
To build the double-basin potential, the reference structure of H1 conformation was directly taken from the Protein Data Bank(PDB code:2GKU),while the structure of H2 conformation was modified from a similar topology formed by sequence Htel25 (5′-TAGGG[TTAGGG]3TT-3′, PDB code:2JSL). Using the Discovery Studio, the 3′-thymine in Htel25 was truncated, and the adjacent thymine was mutated to an adenine nucleotide. Likewise,the 5′-adenine was mutated to a thymine nucleotide. Lastly,the modified structure was equilibrated under 300 K with a 100 ns NPT all-atom MD simulation.
Using the above model, multiple unbiased simulations around the folding temperature of H1 conformation (330 K)were performed starting from an unfolded structure. The staring structure was randomly selected from an ensemble of unfolded structures obtained from a 300 ns conventional all-atom simulation under 600 K.The time step was chosen as 2 fs,and the inverse friction constant in Langevin dynamics was 1 ps.Each trajectory was accumulated to 1010steps.
2.2. The Markov state model (MSM) and the transition path theory(TPT)
MSM is powerful to identify metastable states during the conformational changes of biomolecules. In an MSM,by choosing reasonable features to characterize the conformations and reducing the dimension of conformational space with dimension reduction methods, the space is first transformed into a set of discrete microstates. Then, after selecting a lag time under which jumps between these microstates can be regarded as a Markov chain, a few macrostates or metastable states are identified with the usage of Perron cluster analysis(PCCA+)[89]or other methods. In addition,the transition time between two metastable states is acquired by estimating the mean first passage time(mfpt).
On the basis of metastable states obtained from the MSM,folding pathways can be identified using a TPT analysis. By defining the reactant state and the product state,the probability flux tween microstatesiandj,fi j,is defined as

2.3. Seeding conventional all-atom simulations
In the hybrid atomistic structure-based model, only hydrogen atoms are ignored. Therefore,conformations obtained from structure-based simulations are easily converted to allatom structures. We borrowed a procedure in the construction of an MSM to obtain a set of broadly distributed seeds. When building the MSM, three metrics were used as input features to characterize the conformational changes,including the rootmean-square deviation(RMSD)with respect to the H1 conformation, the RMSD with respect to the H2 conformation and the twelveχdihedral of each guanine nucleotide. In the estimation of RMSD, atoms P, C4′and N1 were used for purine bases,and the atoms P,C4′and N3 were used for pyrimidine bases. Using the time-lagged independent component analysis (TICA)[90]with a lag time of 1000 simulation steps, the space was reduced by retaining 90% of the total kinetic variance. After that,500 microstates were obtained by clustering the conformations using thek-means algorithm. From each of the 500 microstates, ten conformations were randomly selected as starting conformations to conduct conventional allatom simulations.
The DNA force field AMBER OL15 was used in the allatom simulations. Each starting conformation was solvated within a periodic cubic box,and the water molecules were explicitly modeled by the TIP3P model. K+and Cl-ions were added to neutralize the system and to maintain a salt concentration of 100 mM. The electrostatic interaction was treated using the particle mesh Ewald(PME)method with a cutoff of 1.0 nm. The cutoff of the van der Waals interactions was also selected as 1.0 nm. All bonds were constrained using the linear constrain solver algorithm,and the MD time step was set to 2 fs. The Berendsen algorithm was used for both temperature and pressure coupling. The whole system was first subjected to a minimization of 1000 steps, followed by an equilibrium run with an NPT ensemble at 1 atm and 330 K for 2 ns. The last conformation was used for an additional 20 ns NPT simulation at 330 K.Overall,5000 trajectories with a total simulation time of 100 μs were collected to construct the MSM.
3. Results
3.1. Efficiency of the hybrid atomistic structure-based simulations
Examination of structure-based simulations shows that the system hopped among three ensembles, which were determined to be the unfolded state, the H1 state and the H2 state (Fig. 2). This is an indication that the hybrid atomistic structure-based model enabled us to sample a broad range of the folding landscape of Htel24. Consequently, conventional all-atom simulations seeded from these structure-based simulations could also cover a large enough conformational space to investigate the folding mechanism and could be used to estimate the real time scale of the folding dynamics.
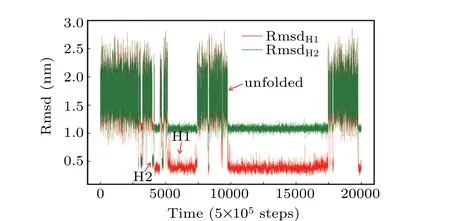
Fig.2.A typical trajectory of the hybrid atomistic structure-based model with a double-basin potential. The RMSDs of all the heavy atoms with respect to the H1 and H2 conformations are colored red and green,respectively.
3.2. MSM
From the conventional all-atom simulations, we built a high-resolution MSM.The RMSD with respect to the H1 conformation,the RMSD with respect to the H2 conformation and the twelveχdihedrals were used as input features. The definition of RMSD can be found in the section of theoretical methods. Using the TICA method with a lag time of 2 ps,the conformational space was projected onto an 8-dimension space by retaining 90%of the total kinetic variance.After that,the conformations were clustered into 300 microstates with the usage ofk-means clustering algorithm. Because the implied time scales reached plateaus after about 3 ns (Fig. 3(a)), this lag time was used to build the MSM.Moreover,since there exists a gap between the zero-based seventh and eighth implied time scales(Fig.3(b)),300 microstates were lumped into nine macrostates using the hidden MSM method,[91]which were labelled as states 1-9.
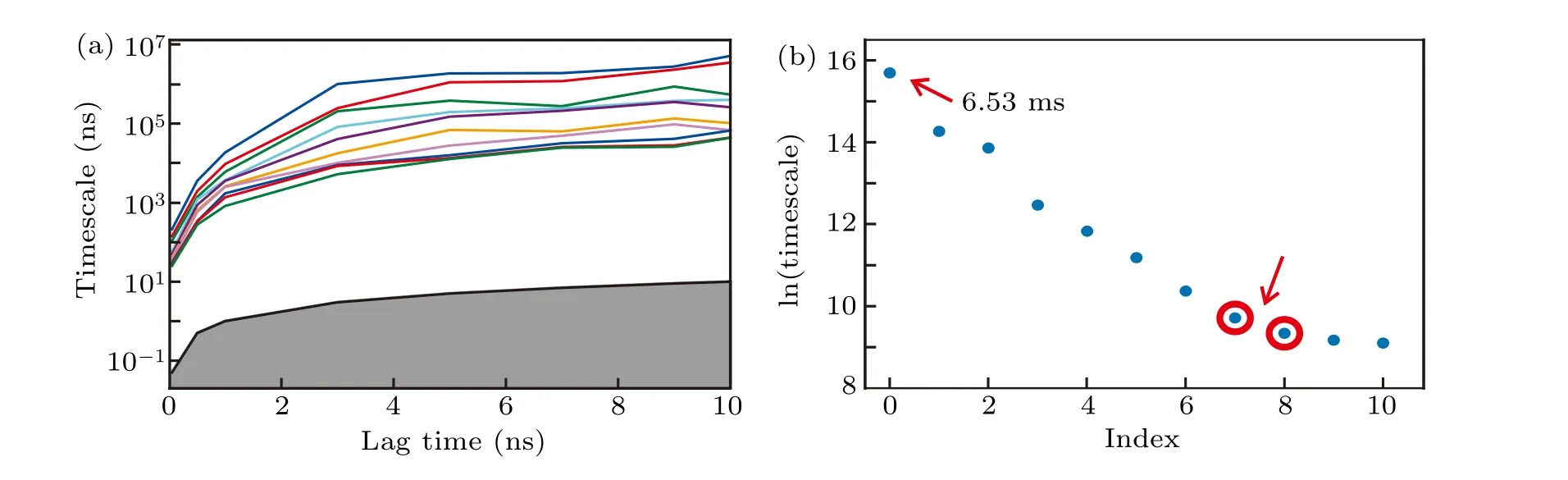
Fig.3. Construction of the MSM.(a)Implied time-scales as a function of lag time. (b)The sorted eigenvalue spectrum estimated from a lag time of 3 ns. The predicted folding time,which is indicated by the slowest time-scale,is approximately 6.53 ms.
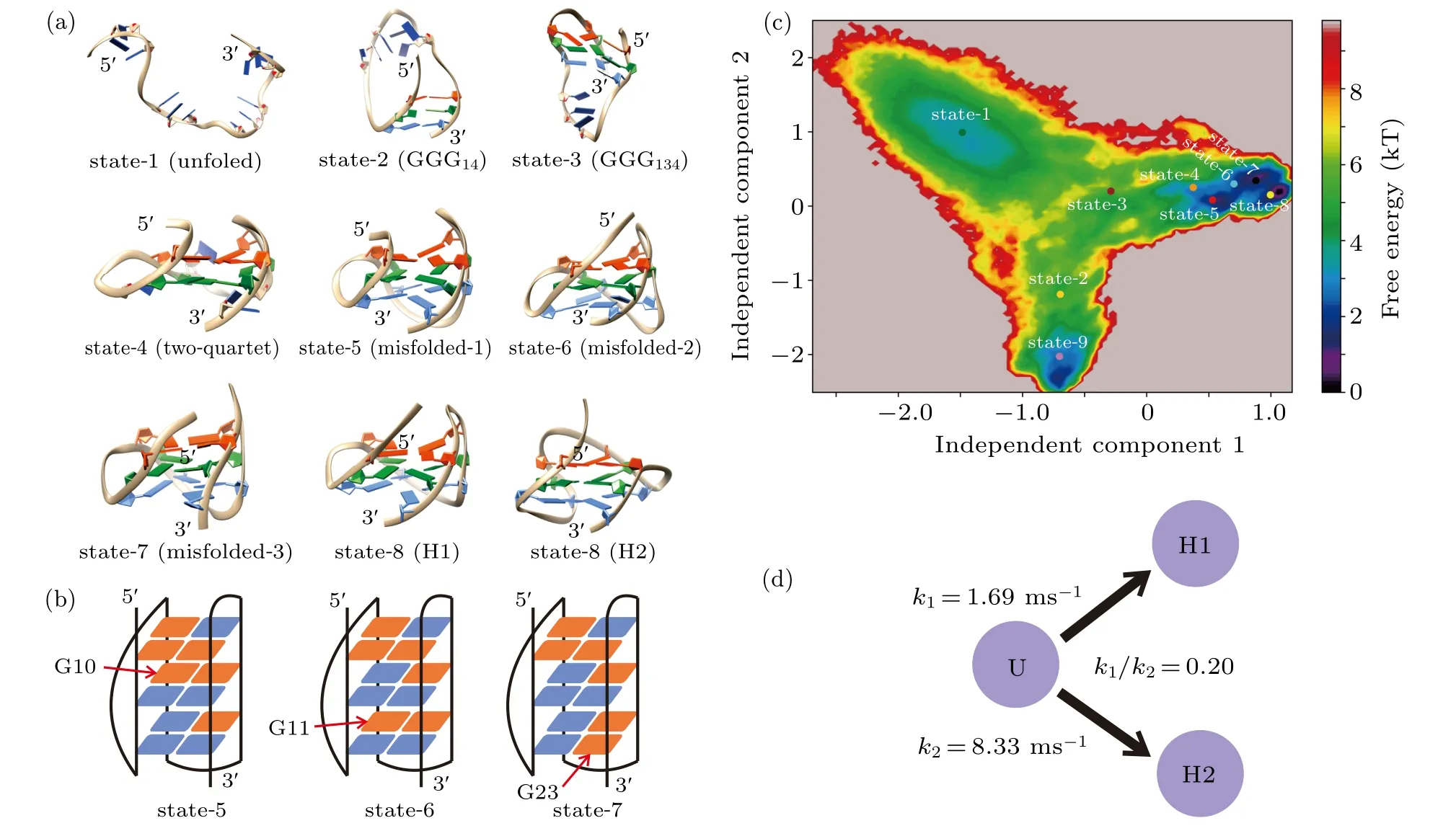
Fig.4. MSM and TPT analysis. (a)A randomly selected representative structure for each metastable state. (b)Distributions of the glycosidic configurations for states 5,6 and 7. Indicated by red arrows are the guanine nucleotides adopting nonnative configurations. The syn and anti glycosidic configurations are colored by red and blue as in Fig.1(b). (c)The free energy profile projected onto the 1st and 2nd slowest TICA components. Also shown are the centers of the nine metastable states. (d) Transition rates from the unfolded state to the H1 and H2 GQ topologies estimated from the TPT analysis.
Figure 4(a) and Figs. S2 and S3 in the Supplementary Material demonstrate the representative structure of each metastable state. State 1 is shown to be highly heterogeneous in terms of structure (Fig. S2). Since it grouped various random coils together,this state was denoted as the unfolded state.States 2 and 3 are partially folded as implied by the structures and can be named as the hairpin state and triplex state, respectively. In state 2 the two terminal G-repeats GGG1and GGG4built a stable hairpin GGG14; in state 3, the G-repeat GGG3docked upon the hairpin and formed a triplex structure GGG134. All the remaining states adopted well-formed fourstranded topologies. It is clear that state 4 is a two-quartet GQ state, which has been suggested to be an important intermediate participating in the formation of three-quartet DNA GQs. Structures of state 5 through state 8 are equivalent to the topology of H1 conformation. However, deep analysis shows that state 5 through state 7 can be looked at as misfolded states(misfolded-1 to misfolded-3),since their distributions of syn/anti glycosidic configurations were incorrect in comparison with the native H1 structure. As shown in Fig. 4(b) and Fig.S4, guanine nucleotide G10 in state 5 took a syn configuration,which should be anti in the native state. Similarly,in states 6 and 7,G11 and G23 adopted the nonnative syn configurations,respectively(Fig.4(b),Figs.S5 and S6). In comparison with the three misfolded states, all guanine nucleotides adopted the native configurations in state 8 (Fig. S7). Therefore, state 8 is characterized as the native state of H1. The competition of the misfolded H1 states and native H1 state is also clearly shown in the free energy profile (Fig. 4(c)). For the last state, i.e., state 9, the representative structure resembles the topology of H2 conformation,and the distribution of glycosidic configurations is demonstrated to be the same as that of the native state of H2(Fig.S8). As a result, state 9 is denoted as the native H2 state.
After identifying the metastable states,the real time-scale transition rate from the unfolded state to the H1 state or the H2 state was obtained by estimating the mftp. In good agreement with the experimental demonstration,the H2 conformation was shown to be kinetically more favorable. It is interesting to note that the ratio ofk1andk2(k1/k2)estimated in this study is on the same magnitude of order with that calculated by the previous simulations (0.20 vs 0.25).[60]Therefore, the all-atom simulations reproduced the folding dynamics discovered by the hybrid atomistic structure-based model.
3.3. Formation pathways of H1 and H2 conformations
The TPT analysis identified multiple formation pathways for the H1 state and one formation pathway for the H2 state(Fig.5). Four pathways,which have probabilities greater than 10%,are discussed for the formation of H1 state. In the dominant pathway(pathway-1),the DNA folded into the H1 state by successively traversing through the ensembles of hairpin GGG14,triplex GGG134and two-quartet GQ state without visiting the misfolded states.
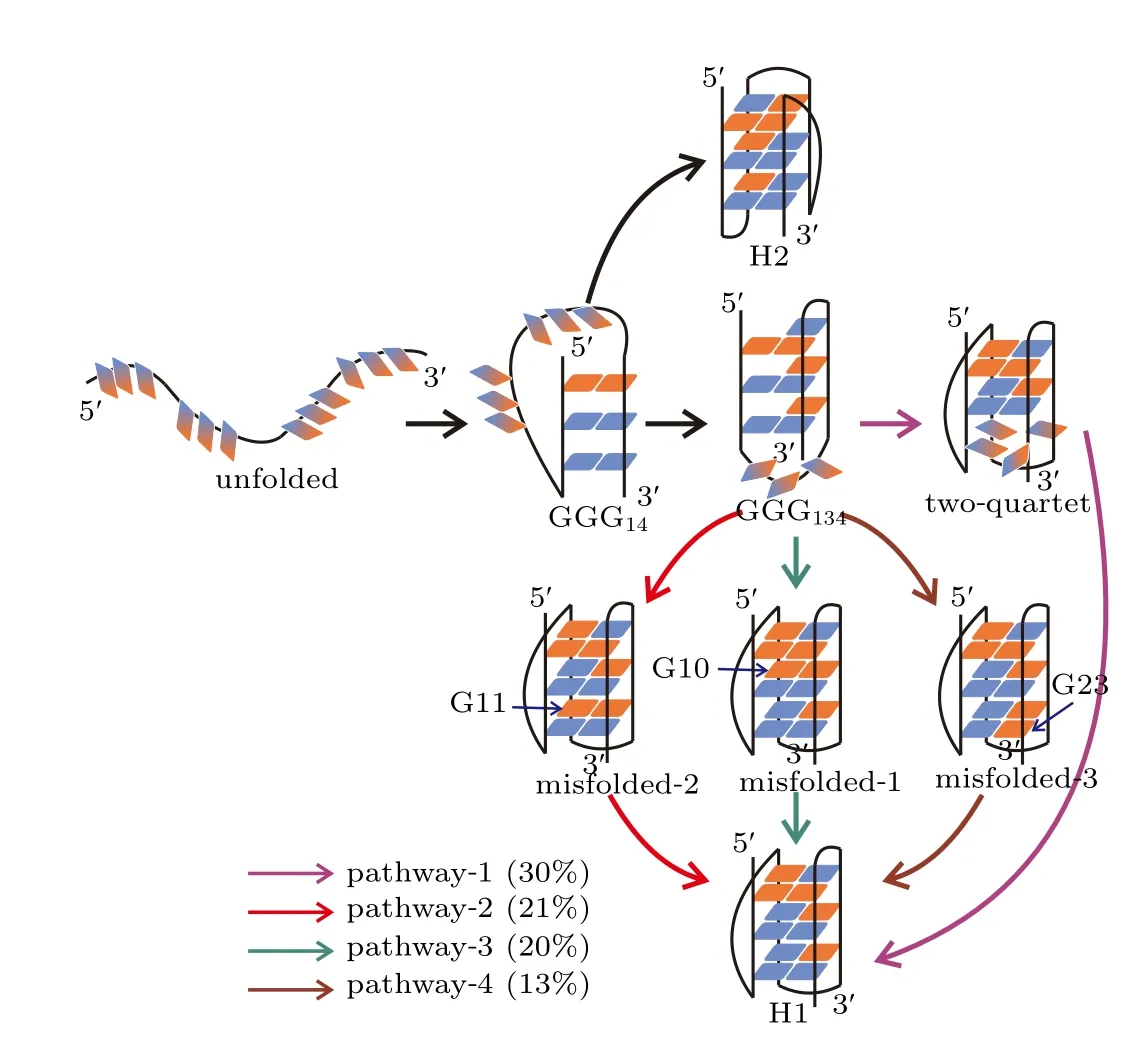
Fig. 5. Formation pathways of the H1 and H2 states. The four major pathways in which H1 forms are colored by violet,red,blue and brown,respectively. Also shown are the probabilities of the pathways. Indicated by arrows are the guanine nucleotides adopting nonnative configurations. The syn and anti glycosidic configurations are colored by red and blue as in Fig.1(b). The gradient color indicates a fluctuating configuration between syn and anti.
In contrast, in each of the other three competing pathways, the system was observed to be trapped in a misfolded state before forming the native structure. For example, in pathway 2, the formation of the H1 state proceeded through the hairpin ensemble GGG14into the triplex state GGG134followed by the formation of the misfolded-2 state in which G11 adopted an incorrect configuration. At the end, the process continued through the nonnative glycosidic configuration reoriented to find the correct pattern. Pathways 3 and 4 are similar to pathway 2 except that different misfolded states were visited. In comparison with the formation of H1 state,the H2 state is demonstrated to form with a higher cooperativity. As shown in the formation pathway of H2 state,the DNA directly folded to the native H2 conformation after going through the GGG14hairpin ensemble.This indicates that the substructures GGG12,GGG23and GGG34can form cooperatively during the formation of H2 state, which has also been observed in the structure-based simulations. It is thus another evidence that the all-atom simulations recovered the folding dynamics revealed by the hybrid atomistic structure-based simulations.
4. Discussion and conclusion
The combined power of the conventional all-atom model,MSM and TPT allows us to complement the folding mechanism of Htel24 revealed by the double-basin hybrid atomistic structure-based model. The relevance of current results to the findings of previous studies is discussed in the following.
The set of all-atom simulations conducted in this study provided an estimation of the real time-scale for the folding dynamics revealed by the hybrid atomistic structure-based model. As shown in Fig.3(b),the folding time of Htel24 under 330 K was estimated to be 6.53 ms. In the absence of direct experimental measurement for the folding time under this temperature, concrete conclusions cannot be made regarding this issue. However, on the one hand, due to the structurebased model favors the H1 and H2 topologies,some potential kinetic traps (such as alternative GQ folds) were eliminated during the simulations. Consequently, the folding time scale estimated here may represent a lower limit and gives a hint of how fast Htel24 may fold if potential competing folds can be avoided by a careful design of experiments. On the other hand,experiments showed that the folding time of Htel24 decreases as the temperature increasing. Since the experimental folding time under 316 K was demonstrated to be on the order of seconds,[45]the time-scale predicted here may be reasonable. This argument can be further verified by an experiment that measures the folding rate.
In comparison with the results of hybrid atomistic structure-based simulations,[60]the all-atom simulations identified more metastable states, including a hairpin state, a triplex state and two misfolded states (misfolded-2 and misfolded-3). Of interest is the triplex state, which was not stable enough to be characterized as a folding intermediate previously. During the structure-based simulations, the folding temperature was chosen with the aim to sample sufficient folding and unfolding events.However,native contacts,which are the most important interactions stabilizing the structures in the structure-based model, could be broken easily under this temperature. As a result,the lifetime of a triplex structure was largely shortened. In contrast, in the conventional all-atom simulations,more factors increasing the stability of the structures can be considered,such as the nonnative interactions and the metal ions. With the formation of these additional important interactions,the stability and lifetime of a triplex state can be significantly increased. The participation of triplex state in the formation of Htel GQ is consistent with several previous studies.[19,21,39,92]However, whether the G-triplex structure can be characterized as a stable folding intermediate of GQs still remains controversial. While many experimental studies support the involvement of G-triplex structures as intermediates in the folding of GQs,computational studies have demonstrated that such structures are unstable and thus cannot be characterized as intermediates. For example,Liet al. detected the existence of a G-triplex state by investigating the folding kinetics of Htel GQ using a magnetic tweezer.[19]However,by performing large-scale atomistic MD simulations, Stadlbaueret al.reported that the G-triplex structures have lifetimes below 1 μs,which is an indication that these structures are thermodynamically unstable.[49]Therefore,the structural stability of a G-triplex structure requires further investigation to reconcile the contradictions between previous studies.
The all-atom simulations also provided another possible explanation for the kinetic preference of H2 conformation. In the structure-based simulations, the H2 conformation was revealed to fold by two steps, while the H1 conformation was suggested to fold by three steps. The higher cooperative folding manner of H2 conformation was believed to be a reason of its kinetic preference. Here, the all-atom simulations also showed the similar high cooperative formation of H2. Moreover, it was revealed that misfolded states adopting inappropriate combinations of syn/anti guanine nucleotides prefer to participate in the formation of H1 conformation. This preference may also be a potential factor inducing the slower kinetic accessibility of H1,since such misfolded structures have been suggested to profoundly influence the folding processes of DNA GQs.[22,49]However, why guanine nucleotides prefer to adopt nonnative syn/anti configurations during the formation of H1 state remains an interesting issue required further investigation. It is the major reason leading to the multipathway formation mechanism of H1 state and one-pathway formation mechanism of H2 state. In addition, two-quartet GQ structures were suggested to merely participate in the formation of H1 state, which is another possible illustration for the different formation mechanisms of H1 and H2 states. In future, we will systematically explore different preference of misfolded states and two-G-quartet GQs during the formation of H1 and H2 states by performing large-scale molecular simulations.
It is worth noting that the role of metal ions was not discussed. The influence of metal ions during the folding of biomolecules is an important issue.[94]Especially, for Htel DNA GQ,monovalent metal ions(typically K+and Na+)can determine the final type of folded structure.[2]However, the hybrid atomistic structure-based model is essentially a highresolution coarse-grained model,in which explicitly modeling of ions still remains a challenge. To our knowledge, there is not any coarse-grained model that can address the question of nucleic acids folding with ions explicitly modeling at this stage. Therefore,we did not consider the influence of ions in the structure-based model and validated our results by comparing with known experimental observations. As discussed above,the structure-based model enabled us to reproduce the experimental observed two-native-state folding thermodynamics and kinetics of Htel24. This is a validation that simulations based on the structure-based model have captured most of the folding properties. It should be noted that, although we performed conventional all-atom simulations with ions explicitly modeled in this study to complement the results of structurebased model, the simulation time is very limit which is only 20 ns for each trajectory. Particularly,the metal ions may not enter to the center of the adjacent G-quartets in such a short time scale. Consequently, using present all-atom simulations to describe the influence of ions is inaccuracy.To alleviate this problem, we are performing additional long time-scale simulations to explore the important role of monovalent ions and the results will be discussed in our future work.
In conclusion, performing large-scale conventional allatom simulations,we have complemented the results of the hybrid atomistic structure-based model for the two-native-state kinetic partition folding of Htel24. The real folding time-scale has been estimated and more details of the folding landscape are acquired. Moreover,the all-atom simulations also provide a possible illustration for the experimentally observed different kinetic accessibility of H1 and H2 conformations. These findings suggest that the multiscale computational framework coupling the multiple-basin hybrid atomistic structure-based model and the conventional all-atom model is a potential useful method to study the complex kinetic partitioning folding process of Htel GQ and the other DNA GQs.

- Chinese Physics B的其它文章
- Numerical simulations of partial elements excitation for hemispherical high-intensity focused ultrasound phased transducer*
- Magnetic-resonance image segmentation based on improved variable weight multi-resolution Markov random field in undecimated complex wavelet domain*
- Dual-wavelength ultraviolet photodetector based on vertical(Al,Ga)N nanowires and graphene*
- Phase-and spin-dependent manipulation of leakage of Majorana mode into double quantum dot*
- Deep-ultraviolet and visible dual-band photodetectors by integrating Chlorin e6 with Ga2O3
- Extended-source broken gate tunnel FET for improving direct current and analog/radio-frequency performance*