
Characterization of Skin Symbiotic Bacteria of Sympatric Amphibians in Southeastern China
2020-12-30 06:59:28XuejiaoYANGXiangleiHOULiWEIYuLIMingshuoQINTianjianSONGandYimingLI
Asian Herpetological Research 2020年4期
Xuejiao YANG,Xianglei HOU,Li WEI,Yu LI,Mingshuo QIN,Tianjian SONG and Yiming LI*
1 Key Laboratory of Animal Ecology and Conservation Biology,Institute of Zoology,Chinese Academy of Sciences,Beijing 100101,China
2 University of Chinese Academy of Sciences,Beijing 100049,China
3 College of Ecology,Lishui University,Lishui 323000,Zhejiang,China
Abstract The fungal infection called chytridiomycosis,caused by Batrachochytrium dendrobatidis (Bd),has given rise to dramatic declines or extinctions of many amphibian species around the world; however,in Asia,this disease has shown a low zoospore load or scant mortality.One potential reason for this may be that certain unique community structures of amphibian skin symbiosis contribute to the outcome of the disease; nevertheless,we know very little about the microbiota in this region.In this study,we used skin swabs of five sympatric amphibian species that have various habitat preferences in Lishui,Zhejiang Province,a place in southeastern China,to explore the skin bacterial communities by using 16S rRNA amplicon sequencing.We detected a total of 1020 OTUs,belonging to 17 phyla,among which Proteobacteria,Firmicutes,Actinobacteria and Bacteroidetes dominated all five host species.Enterobacteriaceae and Exiguobacteraceae and the genera Escherichia and Exiguobacterium belonging to these two families were identified as the most abundant taxa on our focal species.The alpha diversity was significantly lower on the terrestrial species,and also the highly enriched Proteobacteria was found on the terrestrial species,Rana zhenhaiensis,whereas Actinobacteria,Bacteroidetes,Cyanobacteria and Firmicutes were more abundant on aquatic species than on the terrestrial species.Our results suggest that both host species and habitat sites are important factors driving skin microbial diversity and composition and that amphibians in China may harbour unique skin bacterial communities.This study helps elucidate amphibian skin microbial ecology,and with further efforts,the specific mechanism of the interaction between Bd and host amphibians in China could be elucidated.
Keywords bacterial communities,ecomorphology,frog,microbiome,skin
1.Introduction
Vertebrates act as habitats for diverse and complex microbial communities that can contribute to host health or,conversely,be detrimental to the host.This holobiont (host plus symbionts)has aroused widespread concern for the past few years(Rosenberg and Zilber-Rosenberg,2016),such as in studies on different parts of the human body,for example,the intestine(Brookset al.,2018; Vitettaet al.,2018),mouth (Vogtmannet al.,2019),and vagina (Bernabeuet al.,2019; Fettweiset al.,2019).In addition,the skin is the largest organ of the body,along with microbiome on it,is a complex and dynamic ecosystem and is given great importance in human studies (Chenet al.,2018; Byrdet al.,2018).In recent years,the development of next generation sequencing technology allowing such studies about skin microbial symbionts has also been performed in animals,such as mammals (Hoffmannet al.,2014; Olderet al.,2017),birds (Roggenbucket al.,2014; Engelet al.,2018),reptiles(Hydeet al.,2016; Allenderet al.,2018; Walkeret al.,2019),fish(Carda-Diéguezet al.,2017; Minnitiet al.,2017) and,especially,amphibians (McKenzieet al.,2012; Kuenemanet al.,2014; Janiet al.,2017; Medinaet al.,2019).Amphibian skin is among the beststudied systems due to the notorious fungal infection known as chytridiomycosis caused byBatrachochytrium dendrobatidis(hereafterBd) (Bergeret al.,1998; Longcoreet al.,1999; O’Hanlonet al.,2018).
Batrachochytrium dendrobatidishas been linked to dramatic declines of more than 500 amphibian species around the world,amounting for 6.2% of the 8118 described amphibian species,90 of which have been extinction (Scheeleet al.,2019;AmphibiaWeb,2020).An increasing number of studies have found that the skin microbiome of amphibians provides the first line of defence againstBdthrough the production of antifungal compounds such as violacein,indole-3-carboxaldehyde,2,4-diacetylphloroglucinol (DAPG) and prodigiosin (Bruckeret al.,2008a,b; Harriset al.,2009; Woodhamset al.,2018; Madisonet al.,2019).Previous studies on amphibian skin symbiosis have mainly focused on Australia,Mesoamerica and America,where most amphibian decline events have occurred,while this phenomenon has been less frequently reported in Asia (Batailleet al.,2018; Scheeleet al.,2019).Despite the widespread occurrence ofBdin Asia (Gokaet al.,2009; Sweiet al.,2011; Baiet al.,2012;Zhuet al.,2014,2016),there remains a lack of observation of large amounts of individual deaths or amphibian population declines (Olsonet al.,2013; Scheeleet al.,2019).One potential explanation is that there may be certain unique community structures involved in amphibian skin symbiosis that contribute to lowBdload or decreased mortality.Therefore,exploring the characterization and investigation of the environmental correlations of the skin microbiota symbionts of native amphibians in Asian regions is crucial for understanding the natural resistance of these animals toBd.
There are still limited number reports on the skin microbiota symbionts of amphibians in Asia.In mainland China,previous studies have mainly focused onRana dybowskiiin northeastern China; this is an economically important frog species because its oviduct can be used in traditional Chinese medicine.These studies discussed the differences in skin symbiotic bacteria between farmed and wild individuals and seasonal changes in the gut and skin microbiotas ofR.dybowskii(Bieet al.,2019; Tonget al.,2019).Zhaoet al.(2019) cloned the 16S rRNA gene to conduct bacterial community analysis of the skin ofOdorrana grahami,a frog mainly distributed in the Yun-Gui Plateau.Studies on skin symbionts of frogs in other parts of China are still scarce,and most studies have focused on only one species.Previous research has shown that amphibian taxonomy (host species) is an important predictor of the diversity and community composition of skin bacteria(McKenzieet al.,2012; Kuenemanet al.,2014; Bletzet al.,2017a),and geography (geographical regions),ontogeny (life histories)and ecomorphology (habitats) are also important predictors of the skin microbiome (Abarcaet al.,2018; Longoet al.,2015; Bletzet al.,2017b).
Here,we examined the skin bacteria of five sympatric amphibian species from Lishui,Zhejiang Province,a place with no alien amphibian invasions (Liu and Li,2009; Liuet al.,2013) and a high diversity of amphibian species endemic to southeastern China.These five species have various habitat preferences,with three species,namely,Fejervarya multistriata,Hylarana guentheriandMicrohyla fissipes,belonging to the aquatic type,generally inhabiting standing water,and one species,Polypedates megacephalus,belonging to the arboreal type,often living in low shrubs,grasses or agricultural crops and approaching standing waters during breeding seasons from April to June.The final species,Rana zhenhaiensis,is a terrestrial frog endemic to China,with adult animals inhabiting forest grasslands and using standing waters for reproduction only in mating seasons from approximately January to April.Their diverse habitat use provides us with an ideal opportunity to investigate and compare the diversity of the skin bacterial communities of sympatric amphibian species,which will be helpful for future research on the relationship of the skin microbiome withBdprevalence in southeastern China.
2.Materials and Methods
2.1.Sample collectionOur study was conducted in April 2019,and we randomly sampled individuals of each of five species (F.multistriata,H.guentheri,M.fissipes,P.megacephalus,R.zhenhaiensis) from Lishui,Zhejiang Province.The five species are mainly distributed in eastern and southern China and some countries in southeastern Asia.They showed low or noBdprevalence and no evidence ofBd-induced mortality events in both historical specimens from museum and specimens from the wild (Baiet al.,2012; Zhuet al.,2014) (for sample information,see Table 1).Similar toF.multistriata,H.guentheriandM.fissipes,P.megacephaluswas also sampled near water because the sampling time was its spawning season,whileR.zhenhaiensiswas sampled on a hillside,away from the water.We followed standard procedures and captured each individual by hand using new disposable polyethylene gloves and then poured~100 mL of sterile purified water to remove dirt and transient bacteria from the surface of each frog (Beldenet al.,2015).After flushing,we swabbed each frog with a total of 30 strokes,including five strokes on each side of the abdominal midline,the inner thighs and the foot webbings of each hind leg (Vredenburget al.,2010).Swabs were stored individually in labelled sterile Eppendorf tubes in a car refrigerator before they were stored at -20 ℃ in the laboratory for further processing.
2.2.Bacterial DNA extraction and sequencingBacterial genomic DNA was extracted from swabs using the E.Z.N.A.Bacterial DNA Kit (catalogue No.D3350-02; Omega Bio-tek,Norcross,GA,USA) according to the manufacturer’s protocol with minor adjustments in the pretreatment of the samples.We first immersed the swabs in 1mL of 1× TE buffer,vortexed and discarded the swabs,centrifuged the suspensions at 10000×g for 3 min,and discarded the supernatant.Then,we added 100 μl of 1× TE buffer,vortexed the mixture to completely resuspend the pellet and followed the protocol provided by the kit.The extracted DNA was quantified by a NanoDrop (Thermo Fisher Scientific,Wilmington,DE,USA) and diluted to 5 ng/μl using RNase-free water.Then,the hypervariable V4 region of the 16S rRNA gene of symbiotic bacteria was amplified using the barcoded primers 515F (5'-GTGCCAGCMGCCGCGGTAA-3')and 806R (5'-GGACTACHVGGGTWTCTAAT-3') (barcode-515F and barcode-806R) (Knutieet al.,2017).The final amplicon reaction contained 5× FastPfu Buffer (100 mM Tris-SO4,200 mM KCl,50 mM (NH4)2SO4,10 mM MgSO4,10% glycerol)(TransGen,Beijing,China),2.5 mM dNTPs,2.5 U/μl FastPfu DNA Polymerase (TransGen,Beijing,China),20 ng of DNA,10 μM barcode primers and PCR grade water to 50 μl.The PCRs included a denaturing step at 95°C for 2 min; 35 cycles of 95 °C for 20 s,51 °C for 20 s,and 72 °C for 30 s; and a final extension for 5 min at 72 °C.The concentration and purity of the amplification products were tested by a Qubit fluorometer and agarose gel electrophoresis,and then,the amplification products were purified and pooled in equimolar quantities and sequenced by Novogene Bioinformatics Technology Company(Beijing,China) on an Illumina HiSeq platform using a 250 bp paired-end strategy (Caporasoet al.,2012).Raw sequencing data are available in the NCBI Sequence Read Archive database with the BioProject accession number PRJNA613376.
2.3.16S sequences and statistical analysesAfter demultiplexing and trimming the barcodes and primers of each sample,the overlapping forward and reverse paired reads were assembled with Flash (Magoc and Salzberg,2011).The assembled fastq files were processed using the open-source Quantitative Insights Into Microbial Ecology 2 (QIIME 2(version 2018.11)) pipeline (https://qiime2.org; Hall and Beiko,2018; Bolyenet al.,2019).We used default parameters in QIIME 2 (q2-quality-filter plugin) to conduct quality control,and then,the sequences were denoised by Deblur in QIIME 2 (q2-deblurdenoise plugin) (Amiret al.,2017).Sequences shorter than 250 bp were discarded (Bletzet al.,2017c).Subsequently,the chimeras were filtered using UCHIME (Edgaret al.,2011).Then,the rest of sequences were assigned into the sequence variants using the Greengenes database (version 13_8) (McDonaldet al.,2012;DeSantiset al.,2006) through the q2-feature-classifier plugin,and the taxonomic composition at the phylum,family and genus levels was generated based on operational taxonomic units (OTU) annotation.After removing the sequences derived from chloroplasts,mitochondria,archaea and eukaryotes (q2-taxa-filter plugin),we removed the OTUs that covered < 0.005%of the total reads (q2-feature-table-filter plugin) (Bokulichet al.,2013).The number of reads per sample ranged from 21,420 to 154,799,and all samples were rarefied to 21,420 reads to mitigate the effects of uneven sampling (Schlosset al.,2013).
We used Faith’s phylogenetic diversity (PD) (Faith,1992)index to construct the rarefaction curve by randomly subsampling the data at a series of sequence depths to test whether the depth was sufficient to capture the majority of taxa(Supplementary Figure S1).Before using the rarefied dataset to estimate alpha and beta diversity of the skin bacteria by QIIME 2,a phylogenetic tree was created for generating phylogenetic diversity measures such as unweighted and weighted UniFrac distances (Lozupone and Knight,2005) or PD via the qiime alignment mafft,qiime alignment mask,qiime phylogeny fasttree and qiime phylogeny midpoint-root commands.Then,alpha and beta diversity metrics were assigned using the q2-diversity plugin.We estimated the observed OTUs,evenness,Shannon and PD of the skin bacteria to assess differences among samples.Kruskal-Wallis tests were used to compare alpha diversity among different species.Beta diversity was calculated with the Bray-Curtis,Jaccard,unweighted UniFrac (based on presence-absence and phylogenetic distance) and weighted UniFrac (based on abundance and phylogenetic distance) distance metrics and visualized with principal coordinates analysis (PCoA).We used permutational multivariate analyses of variance (PERMANOVA) (Anderson,2005) with 999 permutations to determine the role of the host species in shaping skin bacterial communities.We performed further analyses and data visualization in the R environment,version 3.6.1 (R Core Team,2019).We used the R package“ggplot2” (Wickham,2016) to generate stacked bar charts at the phylum,family,and genus levels to visualize differences in the relative abundances of skin bacterial taxa.Using the same package,we visualized the differences in the alpha diversity of the five species.To examine differentially abundant bacterial taxa between the ectomorphs (habitat types) of samples,we used linear discriminant analysis (LDA) effect size (LEfSe)(Segataet al.,2011) on the Galaxy Web platform.We defined the dividing point as LDA ≥2.0 as recommended in the reference.
3.Results
We used 16S rRNA gene amplicon sequencing on the Illumina platform to examine skin bacterial communities from all 46 swab samples of each individual.After sequence filtration and classification,we detected a total of 1020 OTUs belonging to 17 phyla,with 9 phyla shared by all host species,7 shared by 2-4 species and one found on onlyM.fissipes.Among the phyla,Proteobacteria (56-87%),Firmicutes (7-27%),Actinobacteria (3-11%) and Bacteroidetes (2-5%) were the dominant skin symbiont bacteria of all five host species (Figure 1).At the family level,Enterobacteriaceae was the most abundant inM.fissipes,H.guentheri,F.multistriata,andR.zhenhaiensis(21-50%),whereas Exiguobacteraceae was the most prevalent inP.megacephalus(22%) (Figure 1).At the genus level,Escherichiawas the most abundant inM.fissipes,H.guentheri,andF.multistriata; an unnamed genus in Enterobacteriaceae was the most abundant inR.zhenhaiensis(18-21%); andExiguobacteriumwas the most prevalent inP.megacephalus(22%) (Figure 2).
We observed significant differences in the alpha diversity of the skin bacteria among host species (Shannon,Kruskal-Wallis:H=19.1905,P=0.0007; PD,Kruskal-Wallis:H=17.5626,P=0.0015; observed OTUs,Kruskal-Wallis:H=19.2590,P=0.0007;evenness,Kruskal-Wallis:H=15.2537,P=0.0042).Multiplecomparisons showed that the Shannon index and PD were significantly lower inR.zhenhaiensisthan in other species (Table 2,Figure 3),and the results of the observed OTUs and evenness were similar to the Shannon index and PD results (Table S1).

Table 1 Sampling locations for five amphibian species from Lishui,Zhejiang,China.
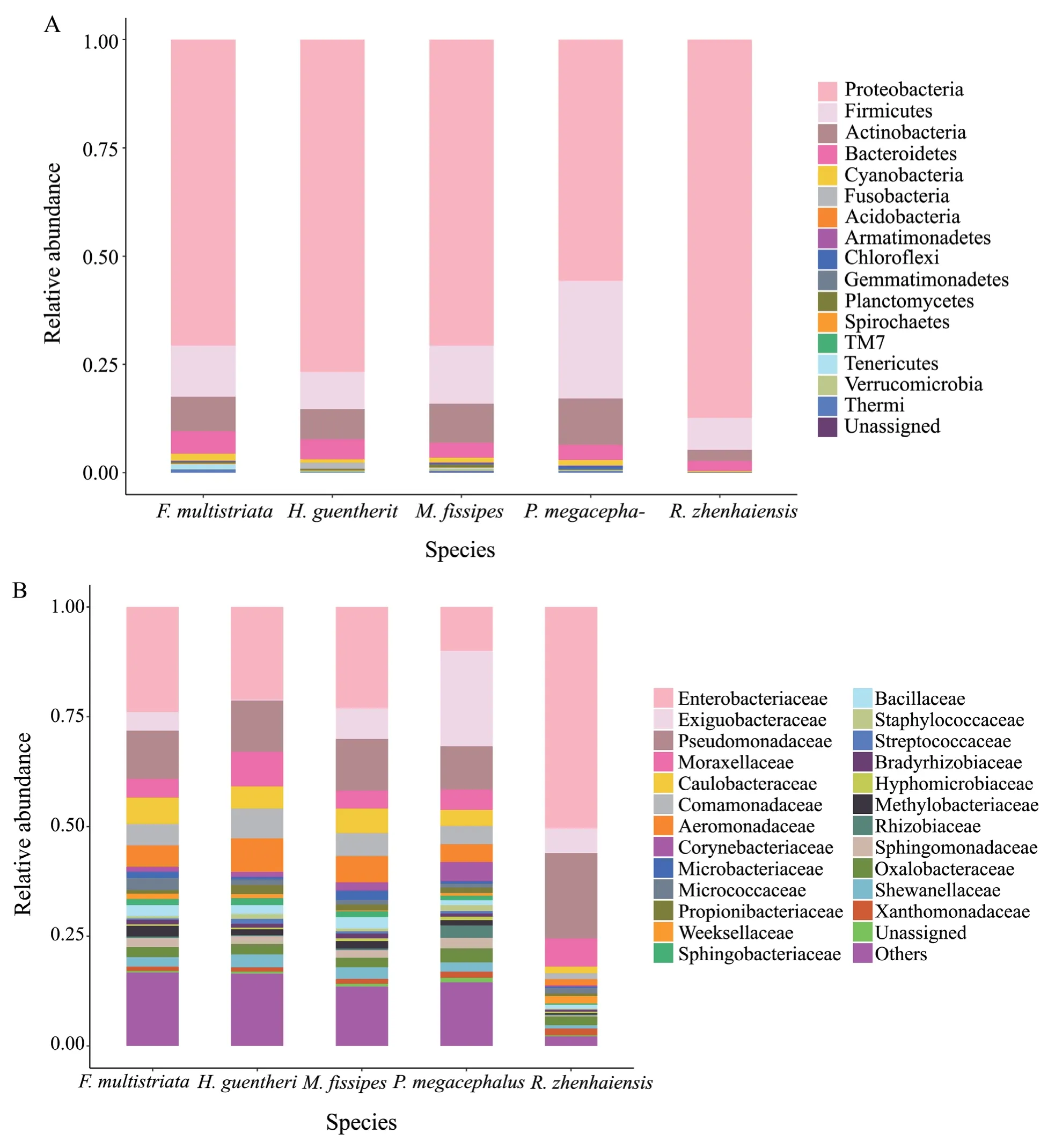
Figure 1 Relative abundance of skin bacterial taxa of five host species.A) Relative abundance of skin bacterial taxa at the phylum level across species.B) Relative abundance of skin bacterial taxa at the family level across species.Taxa with relative abundances < 1%were clustered together.
For beta diversity of the skin bacteria,PERMANOVA analyses showed that the host species indeed had a significant effect on both the bacterial community composition (Jaccard,Pseudo-F=1.4747,P=0.001; unweighted UniFrac,Pseudo-F=1.9046,P=0.001) and abundance pattern (Bray-Curtis distance,Pseudo-F=2.3398,P=0.002; weighted UniFrac,Pseudo-F=3.2569,P=0.001).Pairwise comparisons by PERMANOVA indicated that there were differences in bacterial composition among most species pairs,especially betweenR.zhenhaiensisand each of the other four species (Table 3,Table S2).
Given the unique alpha and beta diversity of the skin bacteria ofR.zhenhaiensis,the only species sampled from land,we further explored the role of habitat type or sampling site type on the skin symbiont of the host using LEfSe to identify specific taxa that varied in abundance in the samples sourced from aquatic and terrestrial environments.Interestingly,Actinobacteria,Bacteroidetes,Cyanobacteria and Firmicutes were highly abundant inF.multistriata,H.guentheri,M.fissipesandP.megacephalussamples from watersides areas; however,R.zhenhaiensissampled from land contained more Proteobacteria than other species (Figure 5).
4.Discussion
In this study,we used skin swabs of five co-habiting species to explore the diversity of the skin bacterial communities by using 16S rRNA amplicon sequencing and to describe and assess the traits,interspecies differences and effects of ecomorphology on skin symbionts of five amphibian species in southeastern China.We found Proteobacteria,Firmicutes,Actinobacteria and Bacteroidetes dominated all five host species,but the alpha and beta diversity and the main taxa at the family and genus level of the skin bacteria were different among host species.Lower alpha diversity and richer Proteobacteria were found on the terrestrial speciesR.zhenhaiensisthan on the four aquatic species,F.multistriata,H.guentheri,M.fissipesandP.megacephalus.

Figure 2 Heatmap of bacterial genera with relative abundances > 0.1% across five host species based on Bray-Curtis distance.
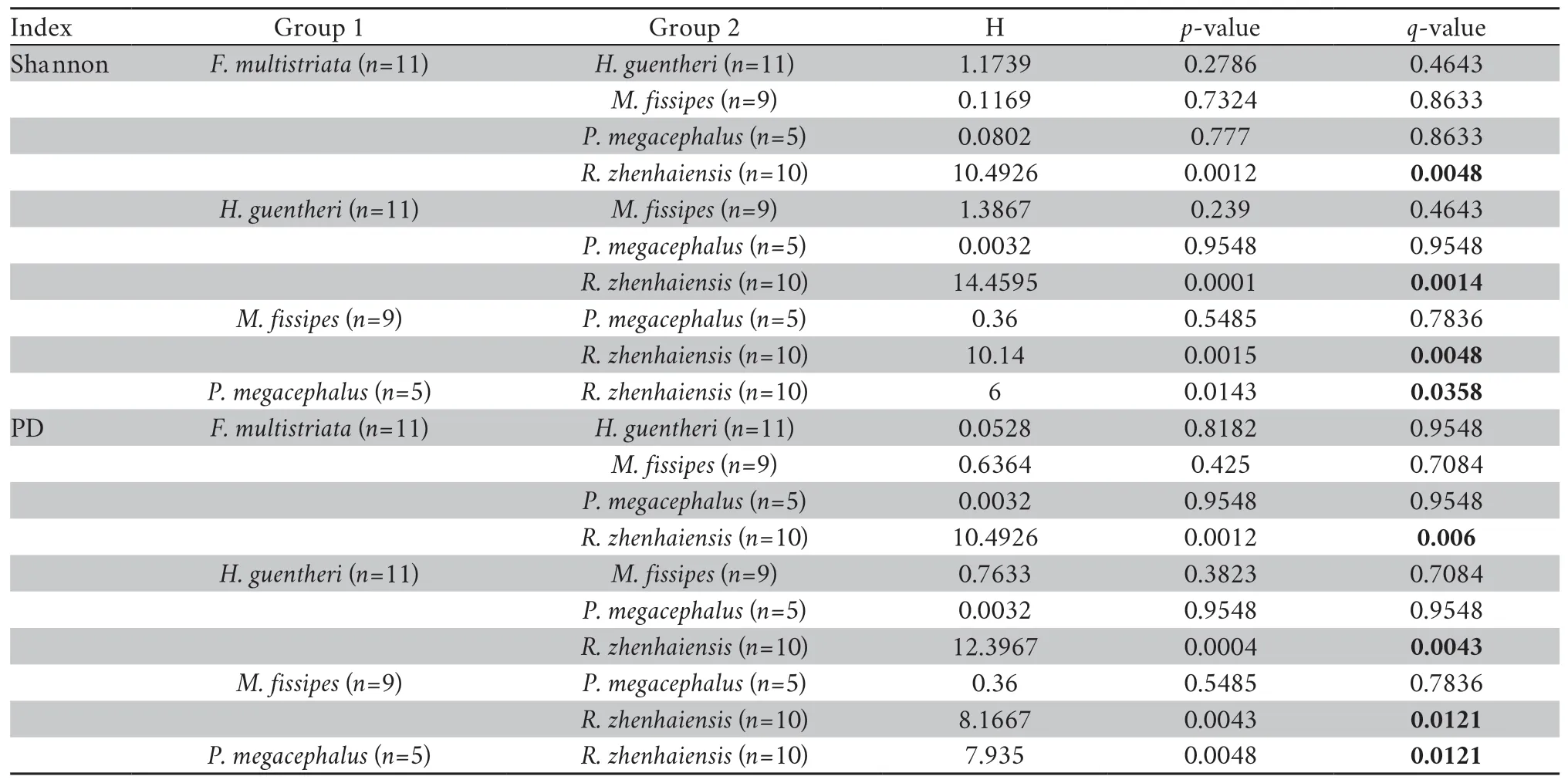
Table 2 Multiple comparisons of species pairs by Kruskal-Wallis tests for Shannon index and PD determination among host species.Boldface indicates significance < 0.05.
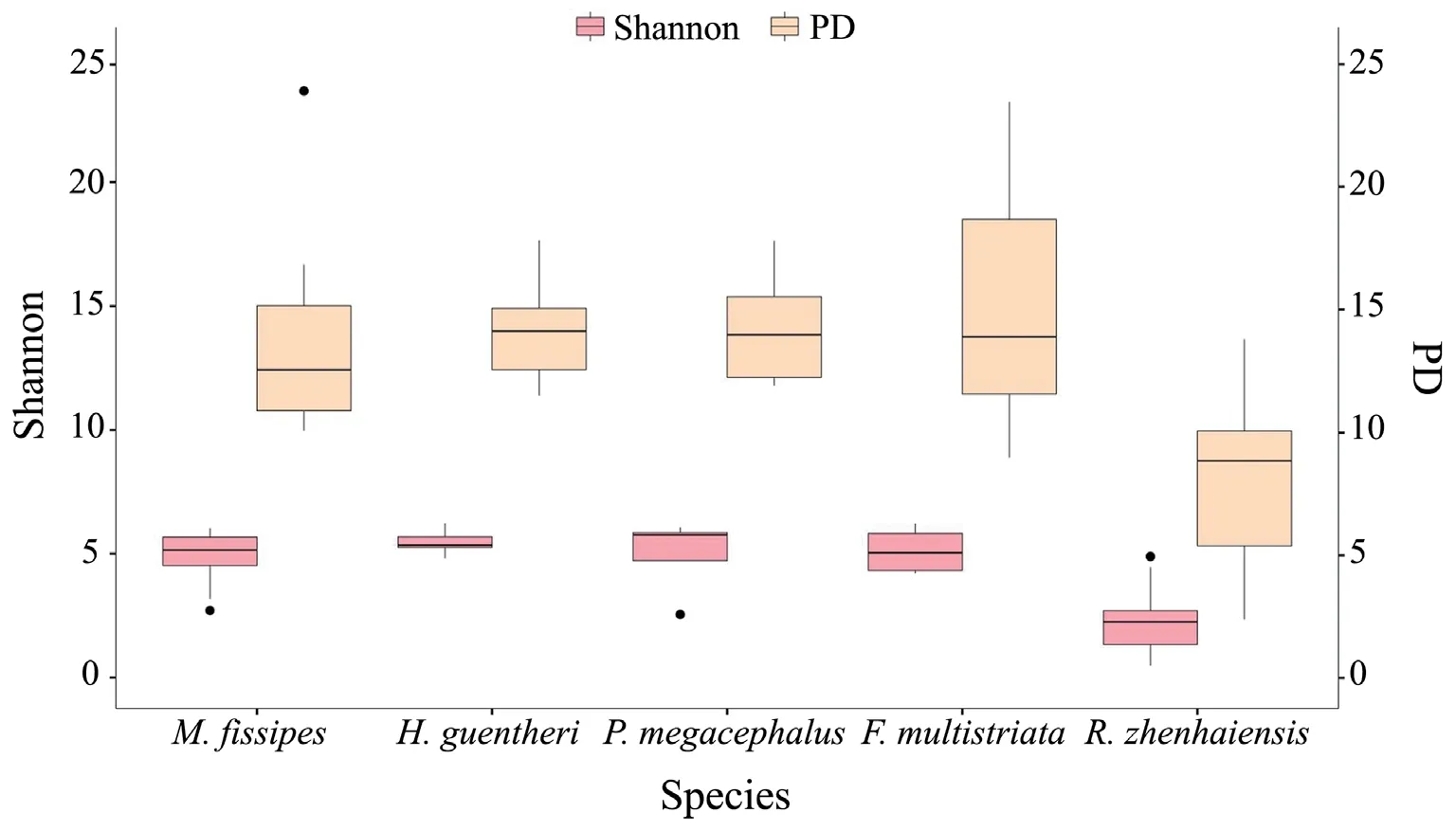
Figure 3 Shannon index and PD of skin bacteria among the five host species.
This is among the first studies to seek the skin symbionts of multiple species and analyse them together in southeastern China,where diverse amphibians are distributed.A detailed understanding of amphibian skin symbionts in different regions of the globe could aid the exploration of measures to mitigate chytridiomycosis (Abarcaet al.,2018).Southeastern China has been proven to be a suitable habitat forBd,as inferred by models (Liuet al.,2013); however,large-scale population declines and extinctions have not been detected by extensive field surveys (Baiet al.,2012; Zhuet al.,2014).One of the possible reasons for this is thatBdoriginated in Asia and has a long coevolutionary history with amphibians in Asia (O’Hanlonet al.,2018).Another important factor is the beneficial effect of the skin microbiome on the amphibian host through microbial species diversity,community composition and assemblage of anti-Bdbacterial taxa,which can produce anti-fungal compounds (Harriset al.,2006; Loudonet al.,2014; Janiet al.,2017; Bellet al.,2018).Considering the close relationship between the skin symbionts and the host,it is of great significance to explore the skin symbionts of amphibians in Asia to understand host health and disease resistance in this region.
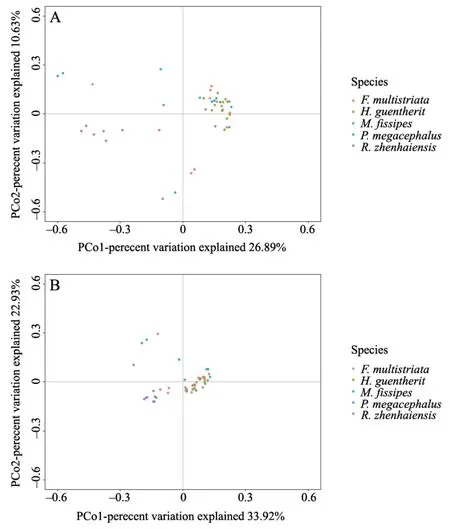
Figure 4 Principal coordinate analysis (PCoA) plots of the beta diversity of the skin bacterial community.A) Bray-Curtis matrices and B) weighted UniFrac matrices of five host species.Each point represents the skin bacterial community of an individual sample.
It has been reported in various studies that host ecology or habitat and/or host taxonomy are crucial drivers of skin microbial composition and diversity in amphibians (McKenzieet al.,2012; Bletzet al.,2017b).We found clear differences in the skin bacteria of the five frog host species that we assessed and pronounced differences in ecomorphology between riparian and terrestrial species in terms of both of the skin microbial diversity and composition.With regard to the Shannon index and PD,the main difference was reflected in ecomorphology; however,contrary to previous studies that showed that terrestrial amphibians have greater bacterial diversity than arboreal or aquatic species (Abarcaet al.,2018;Rebollaret al.,2019),we found that the bacterial diversity ofR.zhenhaiensiswas significantly lower than that of other riparian frogs.Nevertheless,the Shannon index of the skin bacteria ofCraugastor fitzingeri,a terrestrial toad in Panama,exhibited a median value among the riparian,arboreal and terrestrial amphibians (Rebollaret al.,2016).One of the possible explanations for the results is that the environmental conditions,such as temperature,humidity and altitude,varied in the sampling areas.Alternatively,while our study and Rebollaret al.(2016) contained only one terrestrial species,the studies by Abarcaet al.(2018) and Rebollaret al.(2019)lacked aquatic species.Therefore,studies on additional host species inhabiting different environments around the world are needed to test whether the difference in alpha diversity among ecomorphologies is ubiquitous or limited to specific species.Furthermore,unlike the results for the gut microbiota,we found no evidence for strong phylosymbiosis in these skin microbial communities of the five frog hosts,that is,we did not find skin microbial communities parallel with host phylogeny (Brookset al.,2016).This is consistent with the result of Bletzet al.,2017b.In addition to the difference in coevolution between gut and skin with the bacterial symbionts elucidated by Bletz,it is plausible that skin bacteria varied constantly via environmental transmission (Loudonet al.,2014),and probable physical contact between species living in the same or adjacent habitats caused this effect.
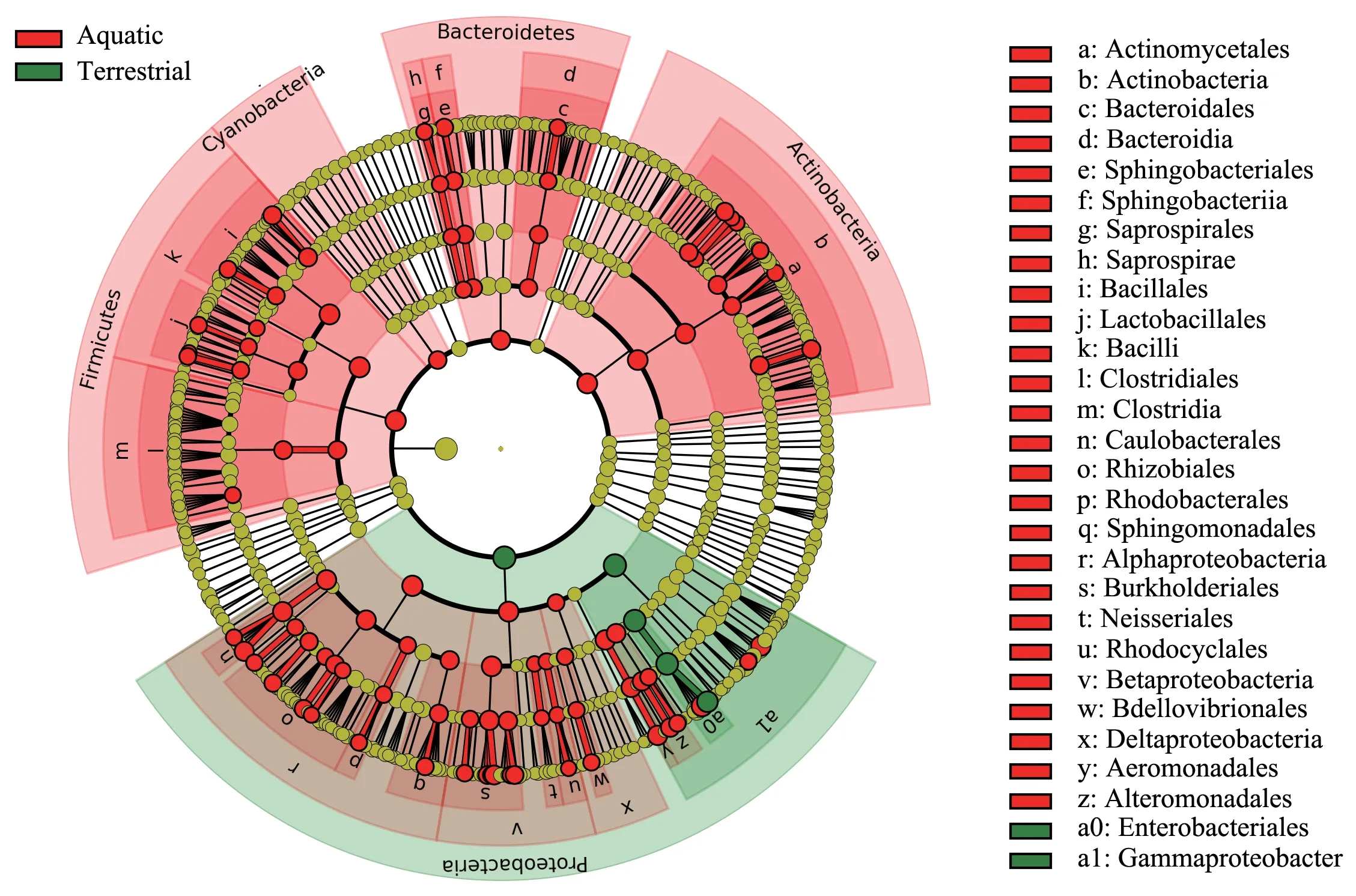
Figure 5 Cladograms showing that the bacterial taxa differ between species sampled from aquatic and terrestrial environments based on LEfSe.
In the present study,we found 17 bacterial phyla on the amphibian skin,among which Proteobacteria,Firmicutes,Actinobacteria and Bacteroidetes were the four dominant phyla,which is consistent with observations in previous studies conducted in both temperate and tropical systems (e.g.,Walkeet al.,2014; Beldenet al.,2015; Bletzet al.,2017b; Jiménezet al.,2019),suggesting that the skin of amphibians may be a selective niche for the colonization of peculiar bacterial taxa (Rebollaret al.,2016; Bletzet al.,2017b).However,the relative abundances of these phyla seemed to vary to some extent across different areas; Proteobacteria was almost the most abundant phylum on amphibians around the world,and the other main phyla may be shifted (Beldenet al.,2015; Bletzet al.,2017b; Abarcaet al.,2018).In this study,Proteobacteria was more abundant on the terrestrial speciesR.zhenhaiensisthan on the other species,which is similar to the results of other studies (Beldenet al.,2015; Abarcaet al.,2018).Proteobacteria was also the most abundant of summer and wild samples of skin microbiotas on the terrestrial speciesR.dybowskiiin northeastern China (Bieet al.,2019; Tonget al.,2019).In southeastern China,Firmicutes was the second most abundant phylum detected on the skin of amphibians,which was different from the results obtained in some tropical countries,such as Panama,Costa Rica and Madagascar (Beldenet al.,2015; Bletzet al.,2017b; Jiménezet al,2019).Apparently,the frogs of temperate regions may have higher Firmicutes abundance than those of tropical zones(Abarcaet al.,2018).
At the family and genus levels,Enterobacteriaceae and Exiguobacteraceae and the generaEscherichiaandExiguobacteriumbelonging to these two families were identified as the most abundant taxa on the skin of our focal species.This result is interesting in light of largely similar studies that showed that the most dominant taxon was Pseudomonadaceae orPseudomonasin different areas (Beckeret al.,2015; Rebollaret al.,2016,2018; Prado-Irwinet al.,2017; Catenazziet al.,2018; Bieet al.,2019).However,Enterobacteriaceae was the most abundant family onMantella aurantiacain Madagascar (Passoset al.,2018).Interestingly,Enterobacteriaceae and Pseudomonadaceae often exhibit high relative abundances together on the skin of many amphibians distributed in many countries,such as Madagascar,Germany,USA,and Australia (Bletzet al.,2017b,c,d; Kearnset al.,2017; Bellet al.,2018).Additionally,a high proportion of inhibitory isolates belonging to Enterobacteriaceae and Pseudomonadaceae was found in the database of culturable anti-Bdbacteria (Woodhamset al.,2015).With regard to the other abundant genus,Exiguobacterium,in our study,also showed high relative abundance in a population of common midwife toads (Alytes obstetricans) (Bateset al.,2018).To date,a total of 17 species have been found to belong toExiguobacterium,and the isolates of this genus have been found to exhibit bioremediation and enzyme production and degradation of toxic substances.Exiguobacteriumis a versatile bacterial genus distributed in a variety of environments (Kasana and Pandey,2018).The high abundance of Enterobacteriaceae and Exiguobacteraceae in this study may indicate the putatively unique anti-Bdfunction among the five sympatric frog species in southeastern China.Of course,further study is needed to verify this hypothesis through bacterial isolation and culture techniques.
To our knowledge,this is the first study to assess differences in the skin microbiota of cooccurring species in southeastern China by high-throughput sequencing.It is important to describe and understand indigenous skin bacterial communities to develop conservation strategies that can be applied locally.Further sampling across a wide area of China is needed to promote the understanding of amphibian skin microbial ecology and to obtain an improved understanding of the particular mechanism of interaction betweenBdand host amphibians in China.
AcknowledgementsThis study was supported by grants from the National Natural Science Foundation of China(31872249 and 31530088).
 Asian Herpetological Research2020年4期
Asian Herpetological Research2020年4期
- Asian Herpetological Research的其它文章
- The Role of Environmental Stress in Determining Gut Microbiome:Case Study of Two Sympatric Toad-headed Lizards
- Adaptive Evolution of the Ventral Scale Microornamentations among Three Snake Species
- Strong Limb Tactics of the Boulenger’s Lazy Toad,Scutiger boulengeri:Inferred from Limb Muscles
- Thermal Biology of Cold-climate Distributed Heilongjiang Grass Lizard,Takydromus amurensis
- The Relationship between Shell Morphology and Crevice Size Affecting Retreat Selection of the Keeled Box Turtle (Cuora mouhotii)
- Spring Voices in Korean Rice Fields:The Effect of Abiotic Variables and Syntopic Calls on the Calling Activity of the Treefrog Dryophytes suweonensis