
CIRCexplorer3:A CLEAR Pipeline for Direct Comparison of Circular and Linear RNA Expression
2019-03-07 07:27:34XuKaiMaMengRanWangChuXiaoLiuRuiDongGordonCarmichaelLingLingChenLiYang
Xu-Kai Ma ,Meng-Ran Wang ,Chu-Xiao Liu ,Rui Dong ,Gordon G.Carmichael ,Ling-Ling Chen ,Li Yang *
1 CAS Key Laboratory of Computational Biology,CAS-MPG Partner Institute for Computational Biology,Shanghai Institute of Nutrition and Health,University of Chinese Academy of Sciences,Chinese Academy of Sciences,Shanghai 200031,China
2 State Key Laboratory of Molecular Biology,CAS Center for Excellence in Molecular Cell Science,Shanghai Institute of Biochemistry and Cell Biology,University of Chinese Academy of Sciences,Chinese Academy of Sciences,Shanghai 200031,China
3 Department of Genetics and Genome Sciences,University of Connecticut Health Center,Farmington,CT 06030,USA
4 School of Life Science and Technology,ShanghaiTech University,Shanghai 201210,China
KEYWORDS Circular RNA;Back-splicing;Linear RNA;Pre-mRNA splicing;Ribo-RNA-seq
Abstract Sequences of circular RNAs(circRNAs)produced from back-splicing of exon(s)completely overlap with those from cognate linear RNAs transcribed from the same gene loci with the exception of their back-splicing junction(BSJ)sites.Therefore,examination of global circRNA expression from RNA-seq datasets generally relies on the detection of RNA-seq fragments spanning BSJ sites,which is different from the quantification of linear RNA expression by normalized RNA-seq fragments mapped to whole gene bodies.Thus,direct comparison of circular and linear RNA expression from thesamegenelociin a genome-widemanner hasremained challenging.Here,weupdatethepreviously-reported CIRCexplorer pipelineto version 3 for circular and linear RNA expression analysis from ribosomal-RNA depleted RNA-seq(CIRCexplorer3-CLEAR).A new quantitation parameter,fragments per billion mapped bases(FPB),is applied to evaluate circular and linear RNA expression individually by fragments mapped to circRNA-specific BSJsites or to linear RNA-specific splicing junction(SJ)sites.Comparison of circular and linear RNA expression levels isdirectly achieved by dividing FPBcirc by FPBlinear to generate a CIRCscore,which indicates therelativecircRNA expression level using linear RNA expression level asthebackground.Highlyexpressed circRNAs with low cognate linear RNA expression background can be readily identified by CIRCexplorer3-CLEAR for further investigation.CIRCexplorer3-CLEAR is publically available at https://github.com/YangLab/CLEAR.
Introduction
Eukaryotic pre-mRNA splicing is catalyzed by spliceosomes to join upstream 5′splice donor sites with downstream 3′splice acceptor sites to produce linear(m)RNAs.Interestingly,downstream 5′splice donor sites can also be linked to upstream 3′splice acceptor sites,referred to as back-splicing,leading to the production of circular RNAs(circRNAs)[1—3].Unlike most mature linear RNAs(including both coding and long non-coding RNAs),circRNAs are covalently closed and lack 3′-end poly(A)tails,resulting in their depletion in poly(A)+RNA-seq datasets.By taking advantage of RNAseq datasets that prof ile non-polyadenylated transcripts and computational approaches that aim to identify fragments mapped to back-splicing junction(BSJ)sites[4,5],a largenumber of circRNAs have been successfully prof iled as being coexpressed with their cognate linear RNAs from the same gene loci[2,3,6—8].Recent studies have shown that the biogenesis of circRNAs is catalyzed by canonical spliceosomal machinery and modulated by bothcis-elements andtrans-factors[1—3,9,10].Importantly,increasing lines of evidence have revealed that somecircRNAsplay important rolesunder physiological and pathological conditions,such as neurogenesis,cancer metastasis,and innateimmune responses,with different modes of action[6,11—14].
Despite these findings,comprehensive characterization of circRNA biogenesis and function has been impeded because the majority of circRNAs are processed from middle exons of genes and their sequences almost completely overlap with those of their cognate linear RNAs except for the BSJ sites[2].Thus,a direct expression comparison of circular and linear RNAs from the same gene loci in a genome-wide manner has remained challenging.The primary obstacle for direct expression comparison is owing to distinct strategies for circular and linear RNA quantification from mapped RNA-seq fragments.In general,RNA-seq fragments that are solely mapped to BSJsites are used to represent circRNA expression,such as by raw or normalized fragment counts(fragments per million mapped fragments,FPM)as shown in Figure 1A(left).On the other hand,RNA-seq fragments mapped to exon bodies and exon-exon splicing junction(SJ)sites are summed up and normalized for linear RNA quantification,such as by fragments per kilobase of transcript per million mapped fragments(FPKM)[15]as shown in Figure 1A(right).Since FPM is unscaled to FPKM,the relative expression levels of most circRNAs are not comparable to those of their cognate linear RNAs when analyzing RNA-seq datasets.
To solve this problem,we have further updated our previously-reported CIRCexplorer[7]and CIRCexplorer2[16]pipelines to version 3 for circular and linear RNA expression analysis from ribosomal-RNA depleted RNA-seq(CIRCexplorer3-CLEAR,or CLEAR for simplicity,Figure 1B).With the CLEAR pipeline,RNA-seq fragments mapped to circRNA-specific BSJ sites or linear RNA-specific SJsites are individually normalized to evaluate circular or linear RNA expression,each in fragments per billion mapped bases(FPB).Unlike using the non-comparable FPM and FPKM values,expression levels of circular and linear RNAs are both quantified by FPB values with the CLEAR pipeline,and thus can be directly compared by dividing FPBcircby FPBlinearto generate a CIRCscore.In this scenario,relative circRNA expression can be evaluated by using linear RNA expression as an expression background, and highlyexpressed circRNAs with low cognate linear RNA expression background can be identified for further functional studies.Parallel analyses further suggest that CLEAR is more reliable for circular and linear RNA expression comparison than other related methods,with economic memory usage and comparable time consumption.
Method
Direct circular and linear RNA expression comparison by the CLEAR pipeline
CLEAR was developed to achieve direct circular and linear RNA expression comparison.Ribo-RNA-seq datasets that prof ileboth polyadenylated linear and non-polyadenylated circular RNAs in parallel are used for precise circular and linear RNA expression comparison.
The CLEAR pipeline includes two main steps:alignment and quantification (Figure 1B). For the alignment,ribo-RNA-seq fragments were first mapped by HISAT2[17](version 2.0.5;parameters:hisat2--no-softclip--scoremin L,-16,0--mp 7,7--rfg 0,7--rdg 0,7--dta-k 1--max-seeds 20)against the GRCh38/hg38 human reference genome with known gene annotations(Figure S1)for subsequent linear RNA quantification analysis.HISAT2-unmapped fragments were then mapped to the same GRCh38/hg38 reference genome using TopHat-Fusion (version 2.0.12;parameters:tophat2 -fusion-search --keep-fasta-order--bowtie1 --nocoverage-search)for subsequent circRNA quantification.
For the quantification,we applied a new FPB value to quantitate linear RNA expression by HISAT2-mapped fragments to SJsitesof the maximally-expressed transcript annotation(Figure S2).The maximally-expressed transcript of a given gene is selected with the highest FPKM value,which is calculated by StringTie(version 1.3.3;parameters:stringtie-e-G)from HISAT2 aligned BAM f ile[18].Fragments mapped to BSJs were retrieved from Top Hat-Fusion as previously reported(version 2.3.6;parameters:CIRCexplorer2 parse-f-t TopHat-Fusion)[16,19]and normalized by totallymapped bases to obtain FPB values for circRNA quantification.
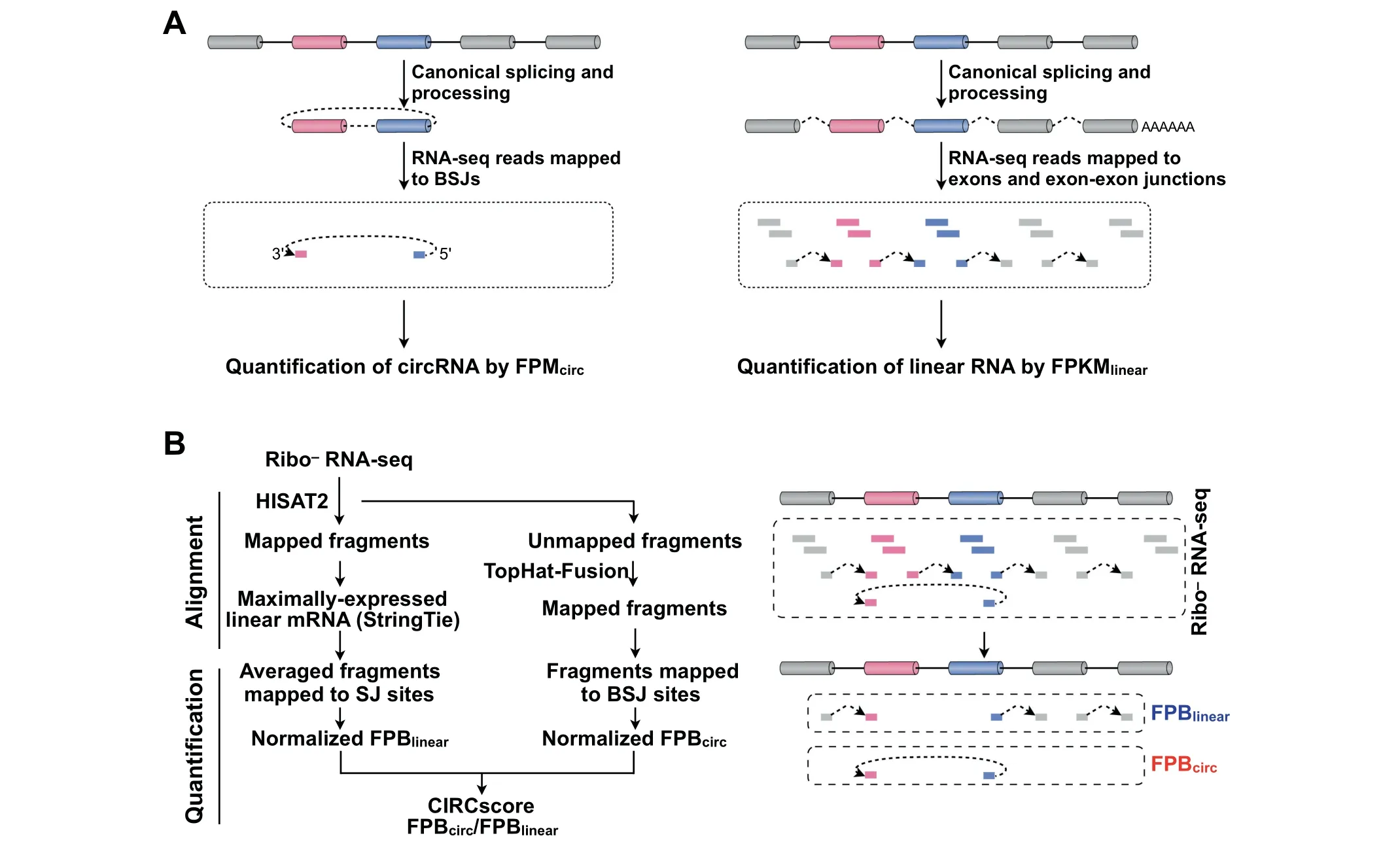
Figure 1 A computational pipeline for direct circular and linear RNA expression comparison
Direct comparison of circular and linear RNA expression is achieved using the CIRCscore value that divides FPBcircby FPBlinear,which represents relative circRNA expression using linear RNA expression as the background.
Flexibility of the CLEAR pipeline
Other aligners,including TopHat2(version 2.0.12;parameter:tophat2-a 6--microexon-search-m 2-g 1)with known gene annotations(Figure S3)or MapSplice(version 2.1.8 with default parameters)with gene annotations(ensGene_v89.txt updated at 2017/05/08)can also be used in the CLEAR pipeline with similar outputs.
In the CLEAR pipeline,comparablecircular or linear RNA expression by FPBs and their direct comparison by the CIRCscorecan beobtained directly from raw RNA-seq FASTQ f iles or processed RNA-seq results,such as CIRCexplorer2 output f iles[16].Please see https://github.com/YangLab/CLEAR for details.
Cell culture
PA1 cells were purchased from the American Type Culture Collection(ATCC;http://www.atcc.org),and maintained in MEMαsupplemented with 10%FBS,1%glutamine and 0.1%penicillin/streptomycin at 37°C in a 5%CO2cell culture incubator.PA1 cells were routinely tested to exclude mycoplasma contamination.
Comparison of FPB with qPCR quantification
Total RNAs from cultured PA1 cells were extracted with Trizol(Thermo Fisher Scientific;Cat No.15596018,Waltham,USA)according to the manufacturer’s protocol.Extracted RNAs were treated with DNase I(DNA-freeTMkit;Thermo Fisher Scientific;Catalog No.AM 1907,Waltham,USA),and reversely transcribed with SuperScript III(Thermo Fisher Scientific;Catalog No.18080044)to produce cDNA and then applied for qPCR analysis.Expression ofACTB,which encodesβ-actin,was examined as an internal control for normalization.Expression of examined linear and circular RNAs was determined from three independent experiments.The primers used in this study are listed in Table S1.
Mapping efficiencies of circRNAs by different pipelines
Three different mapping strategies,including CLEARembedded CIRCexplorer2,MapSplice,and circtools with embedded tool(detect circRNAs from chimeric reads,DCC)[20,21],were applied to fetch fragments mapped to BSJ and/or SJsites in PA1 ribo-RNA-seq dataset[16,22].efficiencies of BSJ-mapped fragments were compared by all three pipelines.Normalized circRNA expression was compared between CLEAR with CIRCscores and circtools with circular over linear ratios(CLRs).
Specifically,for the CLEAR pipeline,fragments mapped to BSJor SJsites and CIRCscores were directly obtained by one single command line:-g hg38.fa-i hisat2_index-j bowtie1_index-G gene.gtf-o out_dir-p 10.For Map Splice pipeline,fragments mapped to BSJsites were obtained by:-p 10--fusion--min-fusion-distance 200--gene-gtf gene.gtf-o out_dir-c chromosomes-x bowtie1_index-1 PA1.For circtools,PA1 ribo-RNA-seq dataset[16,22]were mapped by circtools pipeline with tool(spliced transcripts alignment to a reference,STAR)as suggested at https://docs.circ.tools/en/latest/Detect.html.After a series of reformatting steps,fragments mapped to BSJ or SJ sites were obtained by circtools with parameters:detect@samplesheet-T 10-N-D-an gene.gtf-F-Nr 1 1-fg-G-A hg38.fa-B@bam_f iles.txt.CLRs of circRNAs were finally calculated by dividing BSJ fragments with the mean of SJ-mapped fragments with customized scripts according to circtools.
For the comparison of consumed memories and elapsed time by CLEAR or circtools,ribo-RNA-seq datasets in PA1[16,22]or cortex[23]were used for the analysis with parameters described above.Consumed memories were recorded by linux commandpsevery 20 s.
RNA-seq datasets used in this study
Datasets used for this study include publicly available ribo-,poly(A)+,poly(A)-/ribo-,and RNase R RNA-seq datasets from PA1 cell line[16,22],ribo-RNA-seq datasets of 12 tissues from ENCODE[23](Table S2),as well as ribo-RNAseq datasets of 20 human hepatocellular carcinoma(HCC)samples and their paired normal samples from Gene Expression Omnibus(GEO:GSE77509)[24].
Results
Development of the CLEAR pipeline
The CLEAR pipeline was set up for direct circular and linear RNA expression comparison on a genome-wide scale(Figure 1B).Two characteristic features for circular and linear RNA quantification are applied in the CLEAR pipeline.Similar to circRNA quantification by RNA-seq fragments solely mapped to BSJ sites,fragments that only map to canonical SJ sites by HISAT2 are used for linear RNA quantification(Figure 1B,right).Different from commonly-used FPKM that countsfragmentsmapped to both exon bodiesand SJsites,linear RNA quantification by fragments only mapped to canonical SJ sites is comparable to circRNA quantification by those mapped to BSJ sites(Figure 1B).In addition,fragments mapped to SJ or BSJ sites are normalized by totally mapped bases,rather than by totally mapped fragments,to get FPB for linear or circular RNA quantification(Figure1B,left,Quantification).Direct circular and linear RNA expression comparison can then be achieved with the CIRCscore that divides FPBcircby FPBlinear(Figure 1B,left).
Comparison of FPB with FPKM for linear RNA quantification
To evaluate the accuracy of FPB for RNA quantification,commonly-used FPKM values are obtained from the same HISAT2-mapped results.Basically,HISAT2-mapped results are first converted to BAM format by SAMtools[25].String-Tie[18]is then used to calculate transcript expression by FPKM.Since multiple linear RNAs can be produced from a given gene locus,the average FPB value of fragments mapped to all SJ sites in the maximally-expressed linear transcript is used to represent the expression of this gene in the current study(Figure 1B and Figure S2A,S2B).
With the requirement of FPBlinear>0 and FPKMlinear>0,linear RNA expression,when quantitated by FPBlinear,is highly correlated with that quantitated by FPKMlinearin the PA1 cell line[22](Figure 2A).Indeed,thevalue of FPBlinearis theoretically equivalent to that of FPKMlinear(Figure S2C).Furthermore,FPBlinearis highly correlated with the relative expression of 13 linear RNAs as measured by RT-qPCR in PA1 cells(Figure2B,Table S3).Weobserve a high correlation between FPBlinearand FPKMlinearwhen using different aligners,such as Top Hat2[26]and Map Splice[27],to analyze the ribo-RNA-seq dataset of PA1(Figure S3).Finally,FPBlinearis also highly correlated with FPKMlinearin ENCODE RNAseq datasets from the 12 human tissues examined(Figure S4 and Table S2).Collectively,these findings reveal that FPBlinearis applicable for linear RNA quantification.
Comparison of FPB with FPM for circRNA quantification
As expected,circRNA expression,when quantitated by FPBcirc,is highly correlated with that by FPMcirc(Figure 2C).Experimentally,FPBcircis also highly correlated with the relative expression of 13 examined circRNAs as measured by RTqPCR in PA1 cells(Figure 2D,Table S3).The expression of these 13 circRNAs ranges from~1 to 10 FPB(Figure2D),and their cognate linear RNAs are evaluated above(Figure 2B).
Importantly,compared to commonly-used FPM,FPB is resistant to differences in sequencing lengths and strategies,such as 1×50vs.1×100 or single-endvs.paired-end RNAseq datasets(Figure2E).These resultsare in reasonableagreement with the def initions of FPB and FPM.For example,1 FPB is equivalent to 0.1 FPM for 1×100 bp single-end RNA-seq datasets(Figure S5A)and to 0.2 FPM for 2×100 bp paired-end RNA-seq datasets(Figure S5B).Importantly,in this scenario,FPB can be used directly for crosssample comparison regardless of different sequencing lengths and strategies employed.
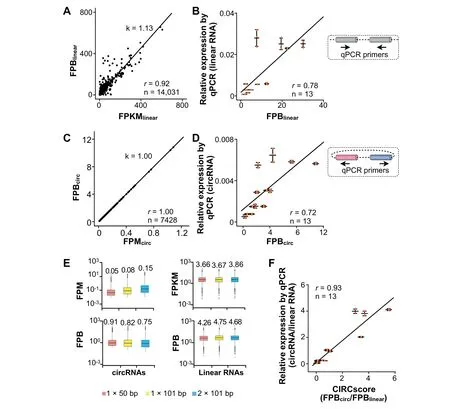
Figure 2 Comparison of FPB with other quantification statistics
Evaluation of relative circRNA expression by CIRCscore
Different from unscaled and non-comparable values of FPKMlinear(for linear RNA expression)and FPMcirc(for circRNA expression),FPBlinearfor linear RNA measurement is comparable to FPBcircfor circRNA measurement.We divide FPBcircby FPBlinearto obtain CIRCscore values,by which expression levels of circular and linear RNAs are directly compared in a genome-wide manner.Importantly,the CIRCscore washighly correlated with theexperimental comparison of circularvs.linear RNA relative expression as measured by RTqPCR in 13 gene loci from PA1 cells examined in this study(Figure2F and Table S3),conf irming that CIRCscoreprovides an additional parameter to evaluate circRNA expression normalized by their cognate linear RNA expression background.
Wefurther compared CIRCscoreby CLEAR with CLR by another previously reported circRNA quantification toolkit,circtools[20].Different from CIRCscore that can be achieved by the CLEAR pipeline directly with a simple command,multiple steps are required to obtain CLR with circtools[20].Importantly,CIRCscore by CLEAR is more accurate than CLR by circtools.For example,CIRCscore of a highlyexpressed circRNA,circCAMSAP-1,is shown as~3.65(Figure3A,blue),which issimilar to RT-qPCR validation with relative expression(circRNA/linear RNA)of~3.82(Figure 3A,gray).However,CLR calculated by circtools with a customized script isabout~0.80(Figure3A,light blue),which is very different from the value derived from RT-qPCR validation.To find out what causes the difference,we performed mapping analysis.It shows that about 126 fragments at the BSJ site ofcircCAMSAP-1 can be identified by the CLEARembedded CIRCexplorer2 pipeline,while only 36 fragments are identified by the circtools-embedded DCC pipeline(Figure 3A),suggesting that DCC could be less efficient for BSJ-mapped fragment calling.The comparison of CIRCexplorer2,Map Splice,and DCC conf irms that DCC is less efficient for circRNA identification(Figure 3).In the ribo-RNA-seq dataset from PA1 cells,356 overlapping circRNAs(with fragments mapped to BSJ≥3)identified by both CIRCexplorer2 and Map Splice failed to be detected by DCC,while only 107 or 99 overlapping circRNAs identified by both CIRCexplorer2 and DCC or MapSplice and DCC were undetected by MapSplice or CIRCexplorer2(Figure 3B),respectively.Among 787 overlapping circRNAs identified by all three pipelines,DCC was also shown to inefficiently call fragments mapped to BSJs in general(Figure 3C).Of note,CIRCexplorer2 and Map Splice are two reliable pipelines for circRNA prof iling[4,28].Taken together,CIRCscore from the CLEAR pipeline is reliable for circRNA normalization using cognate linear RNA expression as background.
Comparison of FPB and CIRCscore in circRNA analysis
circRNAs are generally co-expressed with their cognate linear RNAs and that sequences of circRNAs largely overlap with those of linear RNAs.Therefore,the advantage of using CIRCscoreto quantitate circRNA expression isthat it normalizes circRNA expression to the linear RNA expression background.As shown in the PA1 cell line,among those with FPBcirc≥1,some circRNAs with high FPB values have low CIRCscore values(Figure 4A,blue),possibly due to the high expression of their cognate linear RNAs(Figure 4B).However,other circRNAs with comparable FPB values have relatively high CIRCscores(Figure 4A,red),as their cognate linear RNAs are expressed at low levels(Figure 4C).This observation suggests variable expression patterns of circular and their cognate linear RNAs from different genomic loci.
We further applied CLEAR to evaluate circRNAs in 12 additional human tissues with both FPB and CIRCscore values(Figure 5A and Table S4).Consistent with previous findings[29],circRNAs are more abundant in brain samples than in non-brain tissues.Among all six brain samples examined,circRNAs are more enriched in the cortex,occipital,and diencephalon,but less in the cerebellum,when evaluated by both FPB(Figure 5A,left)and CIRCscore(Figure 5A,right)values.In the six non-brain tissues,circRNAs are enriched in the heart and thyroid at a comparable level to that in the cerebellum.About 10%—20%of circRNAs with FPBcirc≥1 areexpressed at a comparable or even higher level than their cognate linear RNAs,as indicated by CIRCscore≥1(Figure 5A,right),such as in gene loci forcircTPTE2P5andcircPHF7(Figure 5B).Taken together,the identification of highly-expressed circRNAs with high FPBcircand CIRCscore values reveals that some gene loci are particularly favorable for circRNA production(Table S4),and such circRNAs warrant subsequent functional studies.
CIRCscore reduces individual differences
Different from FPB,using CIRCscore to evaluate circRNA expression can reduce individual differences that are caused by RNA-seq samples themselves.For example,compared to paired normal samples,circRNA expression evaluated by the FPBcircvalueisinconsistent in a batch of 20 human HCC samples(GEO:GSE77509)[24].Some HCC samples appear to have generally low circRNA expression;while others,such as samples#11 and#16,appear to have significantly high circRNA expression(Figure S6A).Consequently,it is hard to distinguish circRNA expression differences between HCC and their paired normal samples using FPBcircin these 20 HCC samples(Figure 6A,P=0.99).Strikingly,however,circRNAs are generally lowly expressed in almost all HCC samples when CIRCscore isused to normalizecircRNA expression with cognate linear RNA background (Figure 6B,P=3.59×10-5and Figure S6B).These results suggest that it is important to take cognate linear RNA expression into consideration for circRNA quantification,which can be achieved by the CLEAR pipeline in a genome-wide manner.Taken together,quantification of circRNA expression by CIRCscore helps to eliminate individual differences among paired comparisons,and can therefore be used to decipher the trend of circRNA expression changes under different conditions and for different diseases across RNA-seq datasets.
Discussion
Recently,circRNAs have been widely detected in cell lines and tissues examined by deep sequencing of non-polyadenylated RNAs and using specific computational pipelines for detecting RNA-seq reads/fragments mapped to BSJsites[16,29,30].Due to distinct strategies for circular or linear RNA quantification(Figure 1A),computational pipelines for direct circular and linear RNA expression comparison from RNA-seq datasets have remained challenging.In this study,we have developed CLEAR by applying normalized RNA-seq fragments solely mapped to BSJ or canonical SJ sites individually for circular(FPBcirc)or cognate linear(FPBlinear)RNA quantification(Figure 1B).
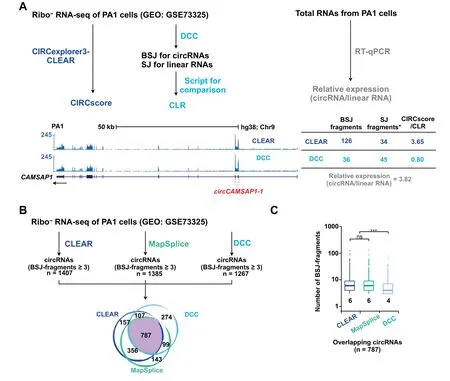
Figure 3 Comparison of circRNA quantification by CIRCexplorer3-CLEAR and other tools
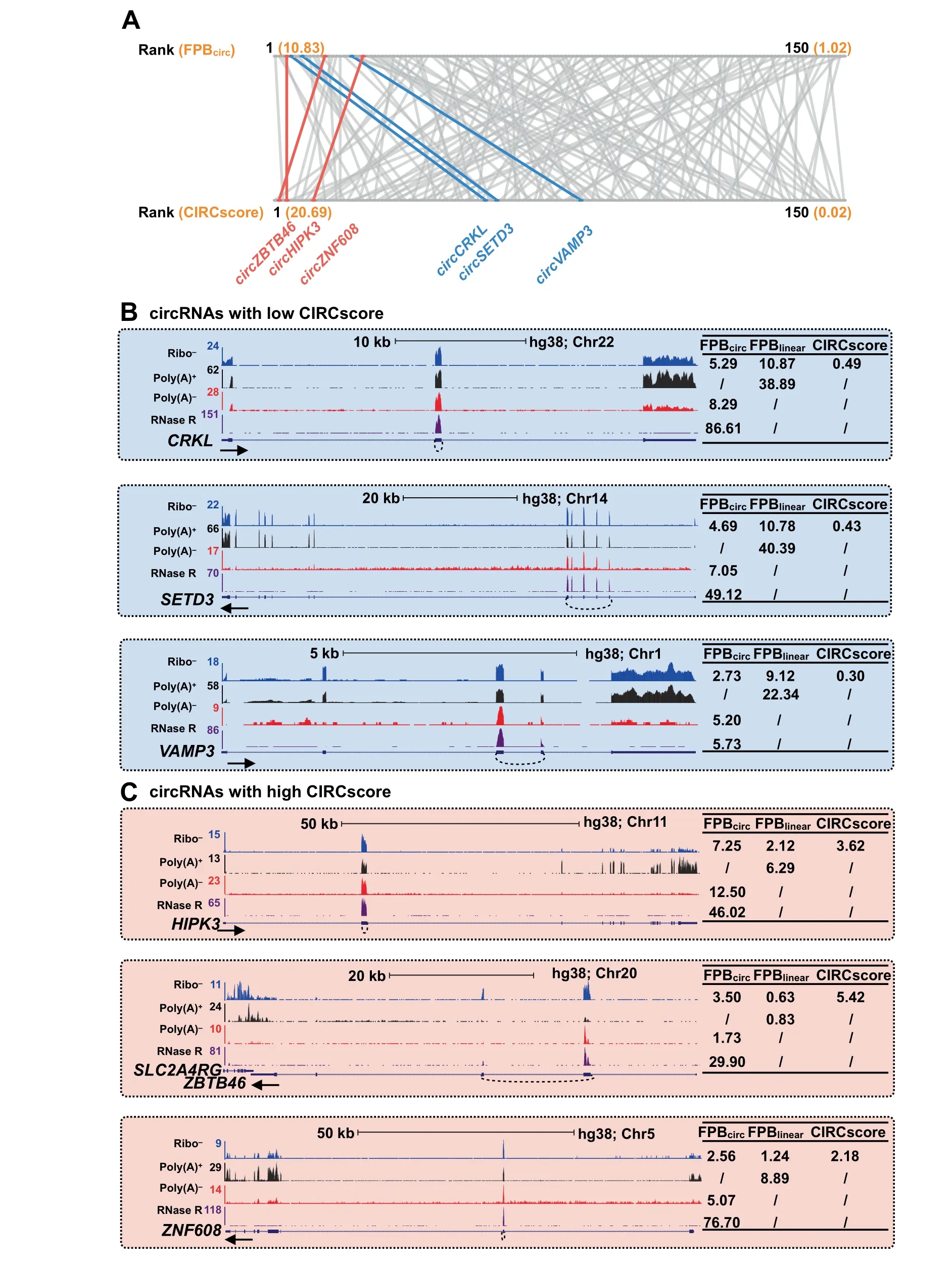
Figure 4 Difference of circRNA quantification by FPB and CIRCscore
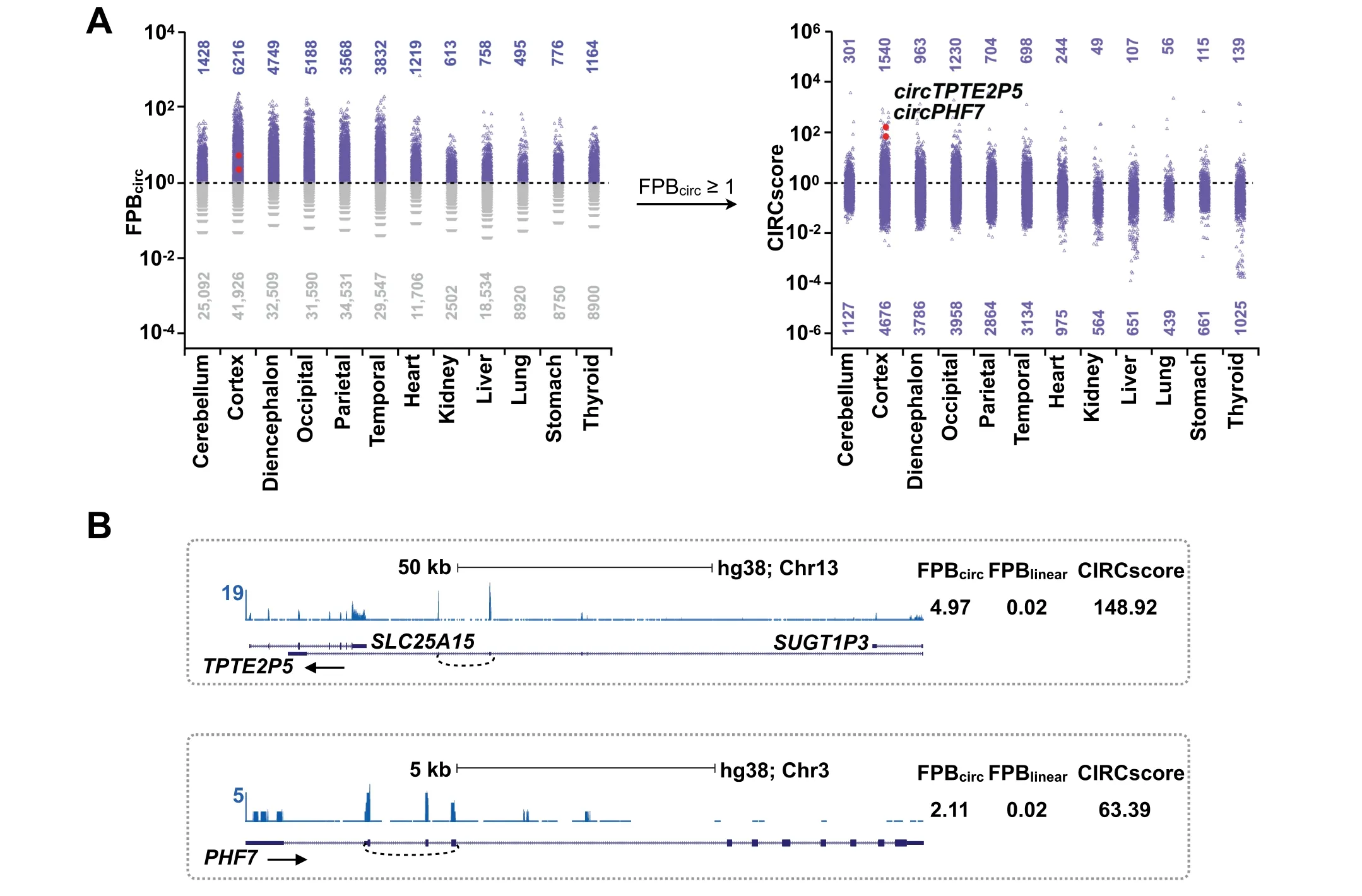
Figure 5 Application of CIRCexplorer3-CLEAR among 12 human tissue samples
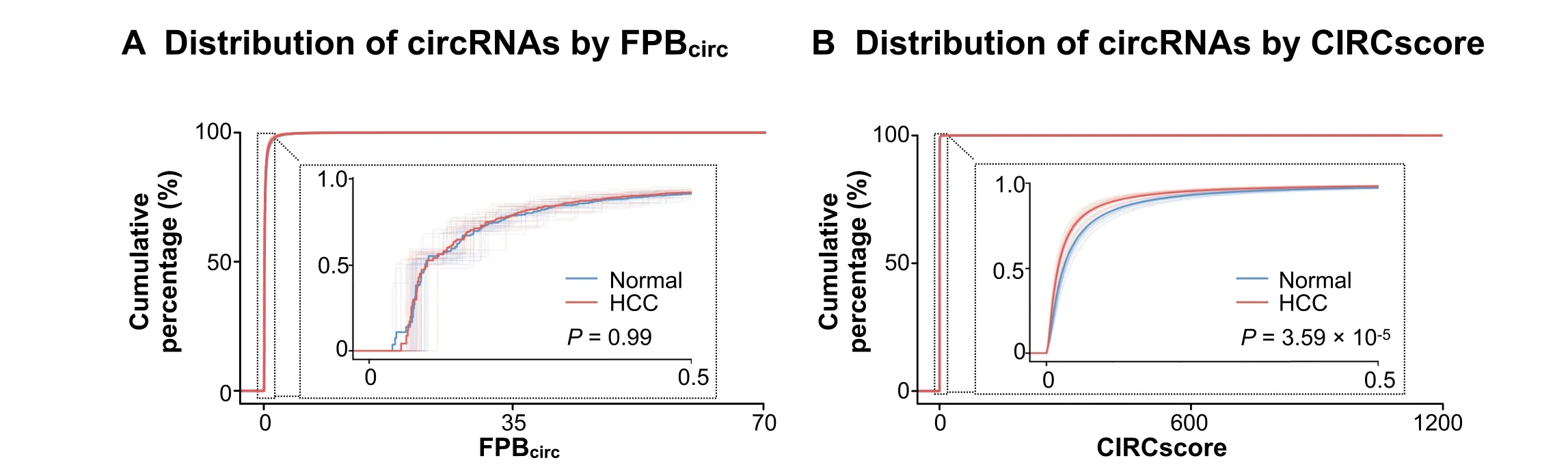
Figure 6 Removal of possible errors/f luctuations and individual differences using CIRCscore quantification
The CLEAR pipeline has at least two advantages in circRNA studies.First,the FPB values are highly correlated with canonical FPKMs for linear RNAs and FPMs for circRNAs(Figure 2),which are unlikely affected by RNA-seq strategies,making cross-sample comparisons feasible.Second,direct comparison of circular and cognate linear RNAs with the CIRCscore not only precisely quantitates circRNA expression relative to normalized linear RNA expression background(Figures 4 and 5),but also eliminates possible errors/f luctuations caused by sample preparation/sequencing differences(Figure 6).This reduces inaccuracies for circRNA quantification and subsequent cross-sample comparison.Compared to other multi-step methods for circular and linear RNA comparison,such as DCC/circtools,the CLEAR pipeline is efficient(Figure 3),memory-economical(Figure S7),easily performed with a single command(Figure S7),and user-friendly due to the application of reliable CIRCexplorer2[4,28].By using cognatelinear RNAsasbackground,CLEAR hasthe potential to allow users to identify highly expressed circRNAs in different biological settings for subsequent functional studies.This is important,because so far it has often been diff icult to identify the circRNAs with the highest expression levels in contexts of interest,or those more highly expressed than their cognate linear RNAs,for functional studies.
It is worth noting that different RNA sequencing strategies have been applied to prof ile circRNAs,including ribo-,poly(A)-/ribo-,and RNase R-treated RNA-seq datasets(Figure 4).Different from poly(A)+RNA-seq datasets that are used to detect polyadenylated cognate linear RNAs,all three types of non-polyadenylated RNA-seq can be used to determine circRNA expression by FPB.However,only ribo-RNA-seq datasets that prof ile both polyadenylated linear and non-polyadenylated circular RNAs in parallel are suitable for direct circular and linear RNA expression comparison by CIRCscore(Figure 4).In contrast,in poly(A)-/ribo-,and RNase R-treated RNA-seq datasets,polyadenylated linear RNAs are largely depleted,which is unsuitable for accurate linear RNA quantification and subsequent CIRCscore evaluation.
Taken together,the CLEAR pipeline provides a comprehensive way to quantitatively evaluate circRNA expression acrosssamplesand to identify highly expressed circRNAswith low linear RNA expression background.
Availability
The CIRCexplorer3-CLEAR pipeline and its application can be downloaded from https://github.com/YangLab/CLEAR.
Authors’contributions
LY conceived and designed theproject.XKM,MRW,and RD performed computational analyses under the supervision by LY.CXL performed experiments under the supervision by LLC.LY,LLC,and GGC wrote the paper with input from XKM and MRW.All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the Strategic Priority Research Program of Chinese Academy of Sciences,China(Grant No.XDB19020104),the National Natural Science Foundation of China(Grant Nos.31730111,31925011,and 91940306),and the Howard Hughes Medical Institute International Program,the United States(Grant No.55008728).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.11.004.
 Genomics,Proteomics & Bioinformatics2019年5期
Genomics,Proteomics & Bioinformatics2019年5期
- Genomics,Proteomics & Bioinformatics的其它文章
- Genomics,Proteomics&Bioinformatics
- MakeHub:Fully Automated Generation of UCSC Genome Browser Assembly Hubs
- VPOT:A Customizable Variant Prioritization Ordering Tool for Annotated Variants
- shinyChromosome:An R/Shiny Application for Interactive Creation of Non-circular Plots of Whole Genomes
- CircAST:Full-length Assembly and Quantification of Alternatively Spliced Isoformsin Circular RNAs
- I3:A Self-organising Learning Workf low for Intuitive Integrative Interpretation of Complex Genetic Data