
Abernethy syndrome in Slovenian children: Five case reports and review of literature
2020-10-23 07:25:42JernejaPecekPetjaFisterMatjazHoman
World Journal of Gastroenterology 2020年37期
Jerneja Pecek, Petja Fister, Matjaz Homan
Abstract
Key Words: Abernethy syndrome; Abernethy malformation; Congenital portosystemic shunt; Liver vascular malformation; Children; Infants; Case report
INTRODUCTION
Abernethy syndrome is named after John Abernethy, a London surgeon who first described this disease entity in the 18thcentury[1]. It is a rare anomaly of the portal venous system, whereby the blood from the splanchnic venous system completely or partially bypasses the liver and is diverted directly into a systemic veinviaa congenital portosystemic shunt (CPSS)[2]. The estimated prevalence of this malformation is 1 per 30000 Live births[3]. Spontaneous closure of a CPSS is possible[2]. The incidence of permanent CPSS is around 1 per 50000. CPSS is defined as intrahepatic if abnormal communications between the branches of the portal vein and hepatic veins or inferior vena cava exist, or extrahepatic if it originates from the main portal vein before dividing or when the portal vein is completely absent[4]. Extrahepatic shunts are commonly accompanied by other congenital anomalies[2]and are further divided according to Morgan and Superina into two types. Type 1 represents complete shunting of the portal venous blood (i.e., end-to-side shunt) with severe hypoplasia or total absence of the intrahepatic tree, and type 2, which exhibit partial shunting (i.e., side-to-side shunt) and where some perfusion of the liver with portal blood remains[5]. If there is a congenital absence of the portal vein, type 1 shunt is identified as type 1A, and if the superior mesenteric vein and splenic vein drain into a common trunk and form a sort of portal vein, which then flows directly into the inferior vena cava, the shunt is type 1B – the one that Abernethy originally described[4].
The clinical significance of CPSS mainly depends on the ratio of blood flow through the shunt[6]. The clinical picture of the shunt can range from an asymptomatic patient with incidentally detected elevated liver enzymes or changes detected on liver imaging[2,7,8], to a symptomatic patient with severe disease presenting with multiple organ involvement[7-9].
Diagnosis is usually made by Doppler ultrasonography (US) of the liver[10,11], commonly in combination with computed tomography (CT) or magnetic resonance imaging (MRI) of the abdomen[8,12,13]. A liver biopsy is necessary to confirm or exclude the presence of venules in the portal triads, which is important for distinguishing between type 1 and type 2 extrahepatic CPSS and guiding treatment. Diagnostic testing for the presence of comorbidities or complications is performed based on the clinical picture[2,7,9].
Treatment depends on the type of shunt and the development of complications[14]. For type 1 CPSS, the only definitive treatment option is a liver transplant, whereas type 2 CPSS and intrahepatic CPSS are amenable to percutaneous or surgical closure[2,14-16]. Unlike extrahepatic shunts, intrahepatic CPSS can close spontaneously in the first year of life[2,7,17,18].
Herein we present five children diagnosed with Abernethy syndrome in the last 15 years at the University Children’s Hospital in Ljubljana. We reviewed their clinical features, radiological and laboratory findings, management strategies and outcomes, which are summarized in Table 1.
CASE PRESENTATION
Chief complaints
Case 1:A six-year-old boy presented with severe, recurrent gastrointestinal bleeding and abdominal pain.
Case 2:A newborn boy developed jaundice after the start of oral feeding following abdominal surgery.
Case 3:A 22-month-old boy was referred to our clinic because of breathing difficulties during a viral infection of the upper respiratory tract.
Case 4:A newborn boy presented because of a vascular malformation on his right leg.Case 5:A 14-year-old girl presented with symptoms and signs of severe osteoporosis.
History of present illness
Case 1:The patient fell ill a few days before admission with abdominal pain and vomiting. His parents also reported that he had been passing dark stools and that he was becoming increasingly more drowsy and somnolent.
Case 2:The patient was born after the completed 38thweek of gestation as symmetrically small for gestational age (SGA) male infant. He developed high ileus and was operated on soon after birth. Intraoperatively, a duodenal membrane was found and duodenotomy with membrane incision was performed and his early postoperative course was mostly unremarkable. We introduced oral feeding on postoperative day 7, when the patient was 9 d old.
Case 3:The patient’s parents reported that he had had exertional dyspnea and frequent tachycardias for several weeks before admission, but that his condition worsened a couple of days prior.
Case 4:The patient was born full-term after a normal pregnancy, with normal birth and length measurements. While still in the newborn nursery, he received 36 h of phototherapy due to hyperbilirubinemia.
Case 5:At the age of nine, she began experiencing recurrent bone fractures. In the seven years prior to admission, she had suffered six bone fractures, which occurred with minor injuries. Her dual-energy X-ray absorptiometry (DEXA) scan showed severe osteoporosis and she underwent diagnostic testing prior to the initiation of bisphosphonate therapy.
History of past illness
Case 1:His previous clinical history was remarkable for a congenital vascular malformation on the left shoulder, cardiomyopathy and increased galactose level, discovered in the neonatal period. Since further testing excluded an inborn error in galactose metabolism and galactose levels normalized without any specific diet, no additional diagnostic testing was pursued at that time. His follow-up echocardiography was also normal.
Case 3:He had suffered a small subdural hematoma after a fall when he was 18 mo old but had been otherwise healthy and developing normally.
Case 5:Her clinical history included an atrial septal defect (ASD) type secundum, which was hemodynamically significant and was therefore surgically closed at fouryears of age.
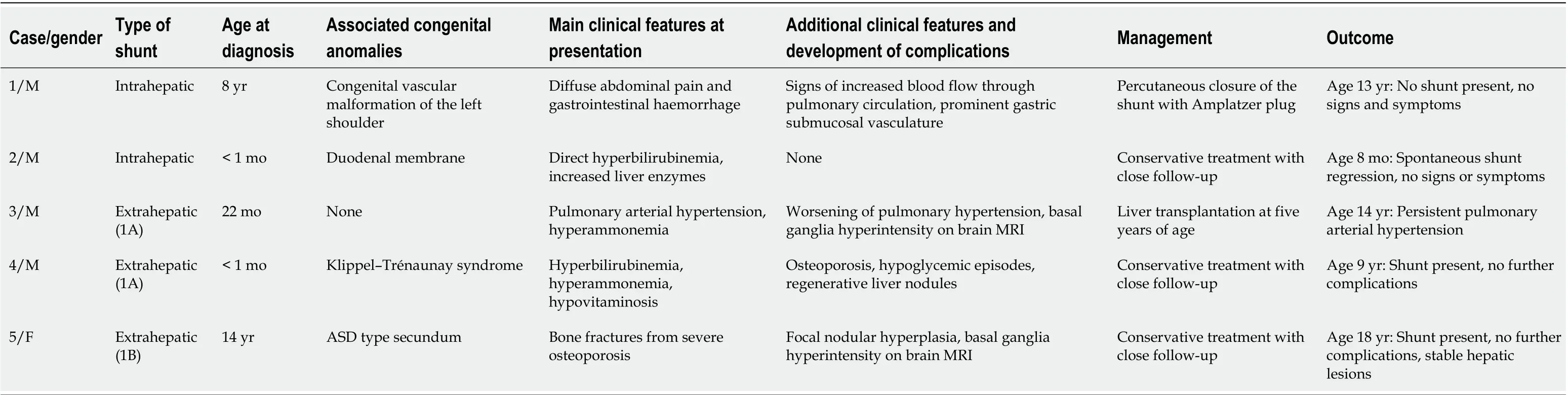
Table 1 Patients' clinical features, management and outcome
Physical examination
Case 1:The patient was awake and responsive, but disoriented. He was pale, with diffuse abdominal tenderness on palpation. Otherwise, his physical exam and vital signs were within normal limits.
Case 2:The main finding on clinical examination was jaundice, accompanied by increasing lethargy and poor feeding.
Case 3:In addition to the signs of acute upper respiratory tract infection, clinical examination revealed peripheral edema and a systolic heart murmur. We also observed a slight tremor of the hands.
Case 4:On clinical examination, an extensive erythematous-livid vascular malformation was noted on his right thigh and lower leg. The patient was also icteric.
Case 5:No abnormalities were discovered in the physical examination.
Laboratory examinations
Case 1:The patient had acute anemia (Hb 79 g/L) and hyperammonemia (ammonia levels 112 μmol/L, normal range 9-33 μmol/L). All other laboratory findings were within normal limits.
Case 2:Blood work-up revealed direct hyperbilirubinemia (133/92 μmol/L) and increased liver enzymes (AST 2.16 μkat/L, normal up to 0.58 μkat/L; ALT 0.94 μkat/L, normal up to 0.74 μkat/L; GGT 0.96 μkat/L, normal up to 0.92 μkat/L). His bilirubin levels continued to rise despite treatment with ursodeoxycholic acid and phototherapy to a maximum of 430/226 μmol/L. When searching for a potential cause, an increased level of galactose in the urine with normal serum galactose and normal GALT enzyme activity in the erythrocytes was found. His serum ammonia was 66 μmol/L (normal range for newborns up to 113 μmol/L) and his coagulation tests were slightly abnormal (INR 1.50).
Case 3:Hyperammonemia was detected. Biochemical investigations otherwise showed good liver function, with normal liver enzymes, increased bile acids and slightly prolonged INR value of 1.69.
Case 4:The laboratory work-up revealed normal liver enzymes, indirect and also direct hyperbilirubinemia, elevated bile acids (neonatal levels of 82 μmol/L, maximum 245 μmol/L at 11 wk of age, normal up to 10 μmol/L), hyperammonemia (maximum ammonia levels 200 μmol/L, normal 9-33 μmol/L), increased levels of alphafetoprotein (1398 kU/L, normal up to 5.5 kU/L), prolonged APTT (49.5 s, normal range 23-42 s) and PT/INR (1.69, normal range 0.90-1.27). He had vitamin A hypovitaminosis (0.19 μmol/L, normal range 0.7-2.8 μmol/L) and his vitamin E and D levels were on the lower limit of normal.
Case 5:Elevated liver enzymes were found (AST 0.99 μkat/L, normal up to 0.52 μkat/L, ALT 0.79 μkat/L, normal up to 0.56 μkat/L, GGT 2.69 μkat/L, normal up to 0.63 μkat/L and AP 4.07 μkat/L, normal up to 1.74 μkat/L). Her vitamin D levels were on the lower limit of normal (25-hydroxyvitamin D 33 nmol/L, normal range 32-165 nmol/L).
Imaging examinations
Case 1:At the time of his first presentation with gastrointestinal bleeding, esophagogastro-duodenoscopy (EGD) showed Mallory-Weiss tears. His initial abdominal US was normal and Meckel’s diverticulum was excluded with scintigraphy. The hyperammonemia was thought to be caused by a gastrointestinal haemorrhage and no further work-up was pursued at that time. The bleeding subsided with symptomatic treatment.
He returned to the clinic after one and a half years because of sudden, severe abdominal pain. At that time, his chest X-ray revealed signs of increased blood flow through the pulmonary circulation and on EGD, prominent gastric submucosal vasculature was observed. Abdominal Doppler US showed an anomalous vascular connection between the wider left hepatic vein and left portal vein (Figure 1). The diagnosis of intrahepatic CPSS was confirmed by abdominal CT with contrast, which demonstrated a direct inflow of the left portal vein into the left hepatic vein (Figure 2). An enlarged venous plexus at the gastric antrum was also noted. Liver biopsy was performed and it showed normal hepatic tissue with venules present in the portal triads.
Case 2:His initial abdominal US showed hepatosplenomegaly without biliary atresia. Later, abdominal US was repeated and Doppler study identified an intrahepatic CPSS with two anastomoses between the branches of the portal vein and the branches of the left hepatic vein. It was estimated that 30% of the portal blood flowed through the anastomoses and therefore bypassed the liver. The hepatic structure was otherwise unremarkable and no pathologic lesions were seen. The diagnosis of an intrahepatic CPSS was confirmed by MRI.
Case 3:On echocardiography, his heart was structurally normal, but his right ventricle was hypertrophied and moderately dilated, with mild to moderate tricuspid regurgitation. He was diagnosed with moderate pulmonary hypertension, confirmed by cardiac catheterization, which showed a mean pulmonary arterial pressure of 45 mmHg. On abdominal US and abdominal MRI, portal vein agenesis was confirmed with a communication between the splenic and mesenteric vein and inferior vena cava. Wedge venography confirmed a total absence of the intrahepatic portal vein. Liver biopsy excluded liver cirrhosis. On brain MRI, basal ganglia hyperintensity was seen, but the EEG did not show diffuse slow waves.
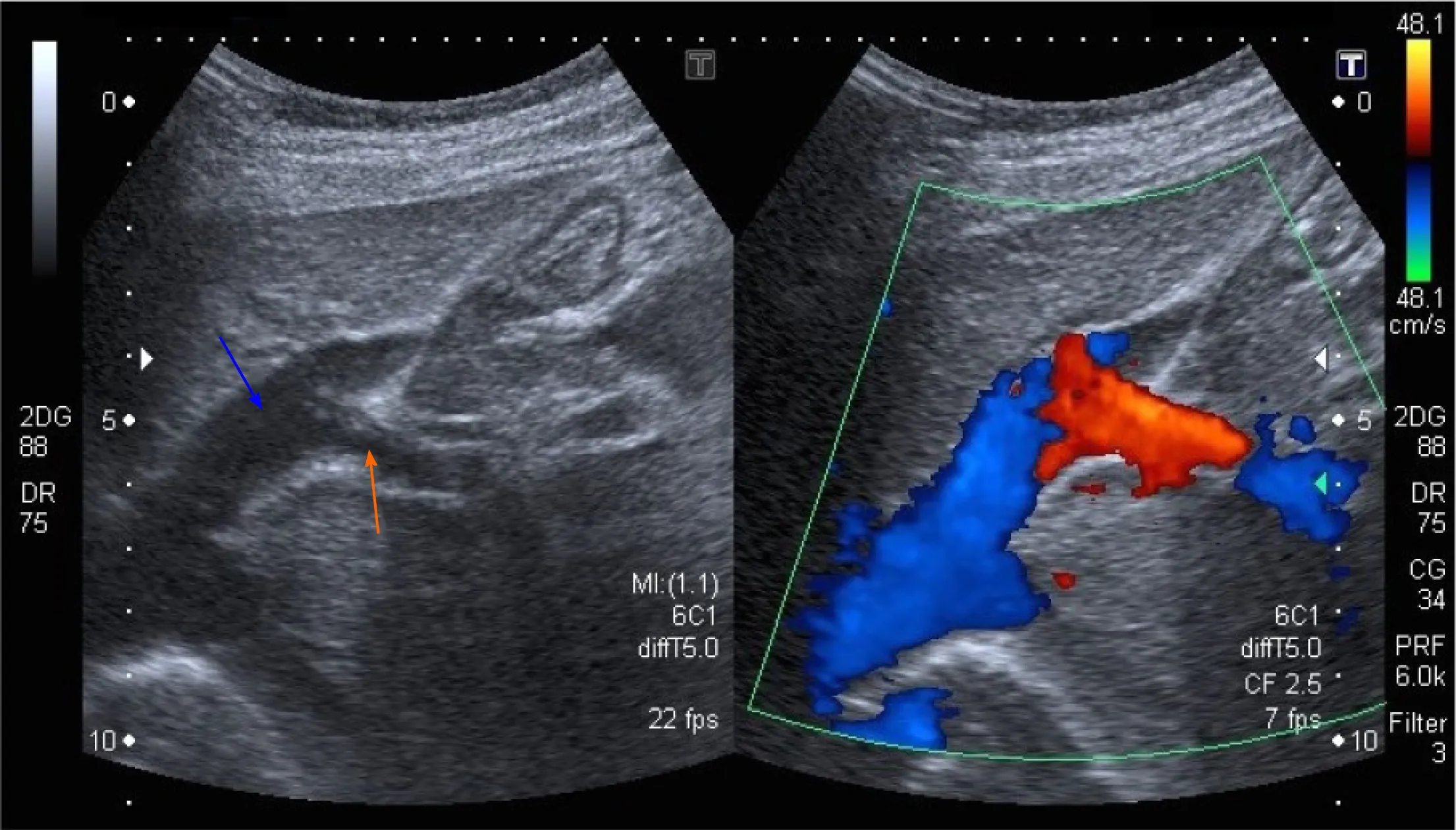
Figure 1 Abdominal ultrasound with Doppler color flow (case 1). An anomalous vascular connection between the left hepatic vein (blue arrow) and left portal vein (orange arrow) is seen.
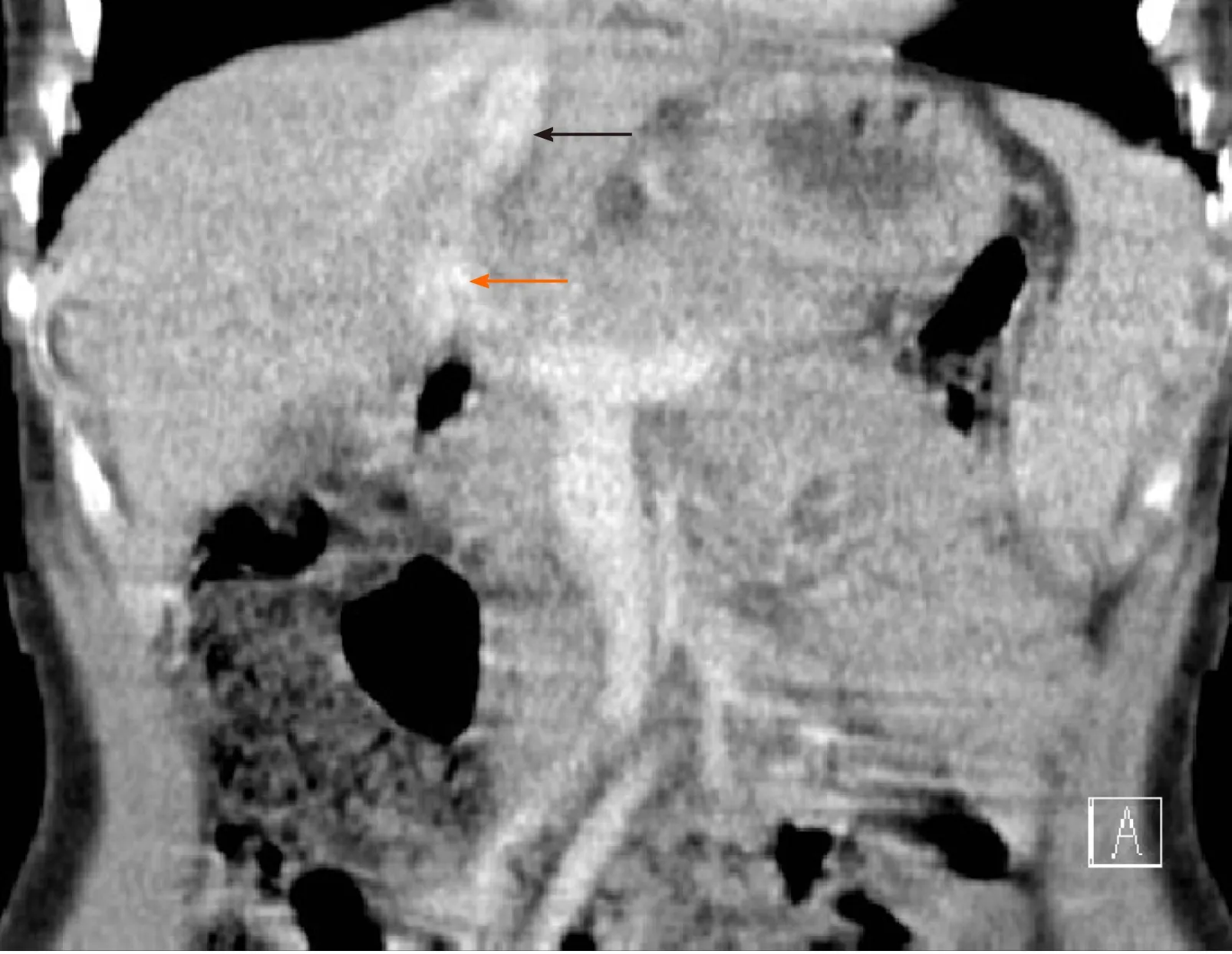
Figure 2 Abdominal computed tomography image of the intrahepatic portosystemic shunt (case 1). The left portal vein (orange arrow) flows directly into the left hepatic vein (black arrow).
Case 4:We performed an abdominal US as a screening test for additional congenital malformations. On Doppler imaging, anomalous abdominal venous vasculature was noted (possible anastomosis of mesenteric vasculature with inferior vena cava) but the exact anatomy was not clear. To obtain a detailed view of his abdominal vasculature, an MRI was ordered, and it displayed a congenital extrahepatic portosystemic shunt without any communication with the intrahepatic portal venous system. The extrahepatic course of the portal vein was not visible. A liver biopsy was also performed and it showed diffuse steatosis without any evidence of liver cirrhosis. His EEG showed minimal diffuse slow electrical brain activity and his echocardiography and chest X-ray were normal.
Case 5:An abdominal US revealed a structurally non-homogeneous tumour formation with central calcinations in the right liver lobe. An abdominal CT was also performed and it showed an anomalous course of the portal vein, which was shorter than normal and drained directly into the inferior vena cava from the anteromedial side, just above the drainage of renal veins – an anomaly typical of CPPS type 1B. The liver was completely perfused solely by the strong hepatic artery (complete arterialization of blood flow). The liver parenchyma was also structurally non-homogenous. MRI of the abdomen showed a combination of adenomas and regenerative nodules (Figure 3) and histologic examination of tumor formations revealed focal nodular hyperplasia. Three months after initial diagnostics, she began experiencing headaches and an MRI of the brain was obtained, which showed globus pallidus hyperintensity (Figure 4). Her serum ammonia levels were normal, however, as was her EEG. No other cause for her severe osteoporosis was found and genetic testing for osteogenesis imperfecta was negative.
FINAL DIAGNOSIS
Case 1
The final diagnosis of the presented case is intrahepatic CPSS with an anastomosis between the left portal vein and left hepatic vein.
Case 2
The final diagnosis of the presented case is intrahepatic CPSS with two anastomoses between the branches of the portal vein and the branches of the left hepatic vein.
Case 3
The final diagnosis of the presented case is type 1A extrahepatic CPSS – complete agenesis of the portal vein.
Case 4
The final diagnosis of the presented case is type 1A extrahepatic CPSS.
Case 5
The final diagnosis of the presented case is type 1B extrahepatic CPSS.
TREATMENT
Case 1
We opted for a percutaneous embolization of his shunt with an Amplatzer plug when the patient was 10 years old. A 16 mm large Amplatzer plug was inserted into a short segment of the left portal vein, just behind the bifurcation. Complete occlusion of the shunt was confirmed by Doppler US before discharge.
Case 2
The patient was treated symptomatically with ursodeoxycholic acid and an aminoacid based formula (Neocate LCP?). Later, when a disorder of galactose metabolism had been ruled out, he was switched back to a normal diet. Due to prolonged INR, he received vitamin K intravenously, after which the coagulation normalized. Exchange transfusion was performed due to severe direct hyperbilirubinemia when he was eight days old.
Case 3
At first, the patient was treated pharmacologically with medication for pulmonary hypertension, intermittent vitamin K applications, ursodeoxycholic acid and lactulose, vitamin D3 and vitamin E. At four years of age, he was started on home oxygen therapy and, one year later, he underwent a liver transplantation.
Case 4
The patient was treated symptomatically with lactulose, dietary protein restriction, ursodeoxycholic acid and fat soluble vitamins.
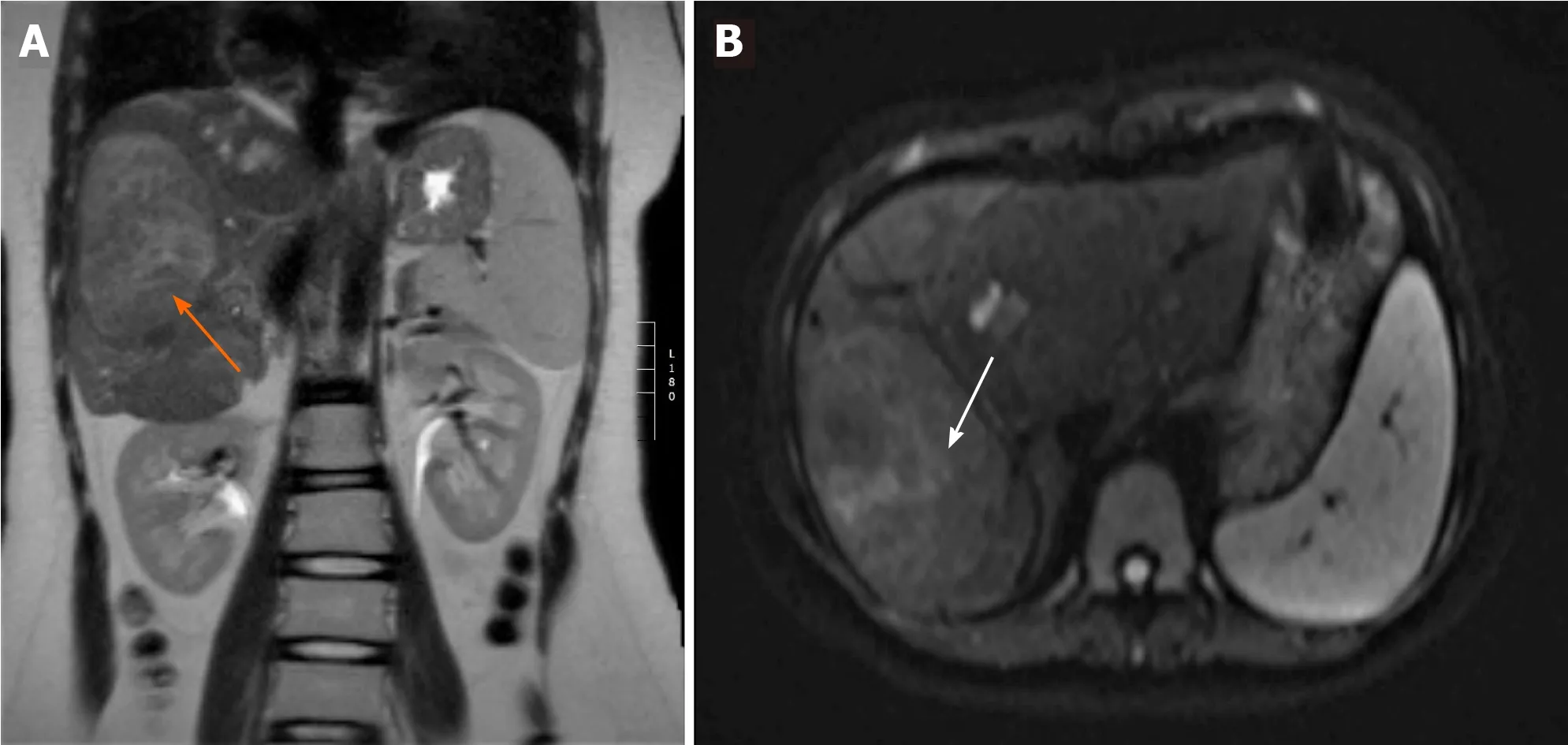
Figure 3 Abdominal magnetic resonance imaging scan demonstrating a lesion in the right liver lobe (case 5). A: The lesion (orange arrow) is slightly hyperintense compared to the sorrounding liver tissue on coronal T2-weighted imaging; B: The same lesion (white arrow) has no restriction of diffusion on axial diffusion-weighted imaging (DWI) sequence - a benign lesion.
Case 5
The patient was treated symptomatically with vitamin D3, calcium carbonate, bisphosphonates, ursodeoxycholic acid and lactulose.
OUTCOME AND FOLLOW-UP
Case 1
In the presented patient, at check-ups, the flow through the shunt did not reappear and, after a couple of months, his right portal vein became significantly more canalised and prominent in comparison with the pre-occlusion state. Since percutaneous closure of the shunt four years prior, the patient has been followed-up regularly and remains asymptomatic. His blood tests and ECG are normal and on EGD, performed 2 years after the closure of the shunt, hypertensive gastropathy was less pronounced than prior to treatment.
Case 2
The patient was discharged from the hospital in good clinical condition and during his follow-up after eight months, a spontaneous regression of the shunt was observed on the abdominal US with color and pulsed Doppler examination. His blood tests were normal and his parents did not report any additional signs; he was developing and growing normally. Ursodeoxycholic acid was discontinued. A follow-up Doppler US is planned at 18 mo of age to see whether the shunt will close completely on its own.
Case 3
Regression of pulmonary hypertension in the presented patient was observed two years after the transplantation. Pulmonary vascular resistance remained elevated, however, so specific pharmacologic treatment was continued. At seven years of age, he underwent treatment for a newly diagnosed Burkitt lymphoma of the small intestine, which occurred during intensive immunosuppressive treatment due to graft rejection. Later, at 14 years of age, during one of his regular echocardiographic examinations, signs of deterioration of pulmonary hypertension were observed, which was confirmed on cardiac catheterization. Further work-up in collaboration with pediatric cardiologists, pulmonologists and hepatologists is planned.
Case 4
When the patient was four years old, we discovered decreased bone mineral density in the range of osteoporosis and calcium carbonate was added to his therapy, while vitamin D3 was switched to its active form (calcitriol). He also began experiencing occasional transient hypoglycemic episodes, which occur about two hours after meals and manifest with sudden tiredness and somnolence. These episodes resolve spontaneously or by ingesting a sugary meal and can be prevented by drinking fruit juices throughout the day.
He is currently nine years old and clinically stable on his medical therapy. On his imaging investigations, we have detected several small hepatic lesions, which have been present and of equal size since he was four years old and have the US appearance of regenerative nodules. His laboratory investigations also did not show any dynamics thus far and he is therefore not on the active transplant list.
Case 5
In the presented patient, regular follow-ups with alpha-fetoprotein, LDH, ammonia, echocardiography and abdominal imaging with Doppler US and MRI are performed. The patient’s osteoporosis is slowly improving on the appropriate therapy, although her mineral bone density is still markedly low. Her liver enzymes also remain elevated. She is at the moment on the non-active transplant list due to the stability of her clinical condition.
DISCUSSION
The first case series of 19 patients was reported in the literature in 1997 by Howard and Davenport[19]. However, the number of reported cases is increasing[2]. In 2013, Sokolliket al[7]identified 316 published cases and, up to 2019, more than 310 cases of just the extrahepatic type of CPSS had been published[8,11]. During a 15-year period (2004-2019), five children were diagnosed with a CPSS at the University Children’s Hospital in Ljubljana. Two of our patients had an intrahepatic CPSS and three had an extrahepatic CPSS – a ratio of 2:3 that is very similar to existing reports[2,7]. Among the children with extrahepatic CPSS, however, two of our patients had type 1A and one patient had type 1B extrahepatic CPSS, which differs from a recent observational, international study by Baigeset al[8], who found that type 1A extrahepatic CPSS were the rarest, representing only 11% of all extrahepatic CPSS in their series.
In our patients, the mean age at diagnosis was four years (range from less than 1 mo to 14 years), which is considerably later than was reported by Kimet al[18], who described that seven out of ten children were diagnosed at an age younger than 1 mo, but in agreement with a larger series by Sokoliket al[7], in which 66% of patients were diagnosed before 12 years of age. One of our patient was female; the others were male. In contrast to that, there is no clear male or female preponderance in patients with CPSS reported in the literature, with 56% males and 44% females affected, but intrahepatic shunts were previously reported to be more common in male patients[2,7].
CPSS, especially type 2 extrahepatic shunts, frequently occur in conjunction with other congenital malformations[2,4,16], which was also true for four out of five patients described. Two patients had congenital malformations of the peripheral vasculature: The patient with an intrahepatic CPSS had a vascular malformation on the left shoulder and the patient with type 1A extrahepatic CPSS had a vascular malformation on his right leg, diagnosed as Klippel–Trénaunay syndrome. The girl with type 1B extrahepatic CPSS was born with ASD type secundum and the second patient described had a congenital duodenal membrane. Whereas congenital cardiac anomalies are found in 20%-30% of CPSS patients[7,8], the association with Klippel–Trénaunay syndrome, which was actually the clue for the discovery of CPSS in one of our patients, has rarely been described[20-22].
Clinical features of CPSS can be divided into several types according to their pathophysiology.
Deficient nutrition of the liver due to a lack of blood flow may cause intrauterine growth restriction[23]. Intrauterine growth restriction affects up to 50% of children with CPSS[2], but it was present in only one of our patients, in whom the lack of portal venous blood flow also caused anoxic-ischemic neonatal cholestasis. Neonatal cholestasis can be a part of the clinical picture and has been described in 24 out of 265 clinical cases of CPSS[2]. Moreover, in 10 of them it was an initial presenting sign that prompted further diagnostic work-up, similar to our case. Searching for a CPSS should therefore be included in the neonatal cholestasis work-up.
Due to the diversion of metabolites and vasoactive mediators from the splanchnic venous system directly into systemic circulation, blood galactose level and blood ammonia may be elevated and portosystemic encephalopathy, hepatopulmonary syndrome or pulmonary arterial hypertension with congestive heart failure might develop. Neonatal hypergalactosemia with normal enzyme activities was detected in two of our patients. According to the published literature, hypergalactosemia is present in up to 70% of newborns with CPSS[2]and vice versa – data show that around 60% of newborns with persistent galactosemia without enzyme deficiencies have CPSS[24]. A CPSS can also lead to hyperammonemia, present in all but one patient from our case series, as well as portosystemic encephalopathy, with a spectrum of different neurological manifestations – from episodes of lethargy or irritability with agitation, to intellectual disability and behavioral problems[2,7]. Typical of portosystemic encephalopathy, but sometimes detected with portosystemic shunting alone, are slow waves on EEG, a minimal degree of which were present in one patient during infancy, while his hyperammonemia was well-controlled on conservative treatment, and a high signal intensity in the globus pallidus on brain MRI, present at the time of diagnosis in two of our patients. This change is thought to be related to hypermanganesemia[2,25]. Takamaet al[26]previously reported on a case of a 1-year-and-7-month-old girl with two intrahepatic portosystemic shunts, who had hypermanganesemia and abnormal globus pallidus hyperintensity which disappeared after the treatment with a right hepatectomy. Levels of serum manganese have not been measured in our patients.
Another common complication of CPSS is porto-pulmonary arterial hypertension, which can occur in children of all ages and with all anatomical types of shunt and can frequently be a first presenting sing of CPSS, as we described in case number 3. Because it can lead to right heart failure and death, screening by clinical history, physical examination and echocardiography are necessary in all children with CPSS, and significant porto-pulmonary hypertension is one of the indications for surgical treatment of CPSS[2,16,27].
One of our patients (case 4) also began experiencing hypoglycemic episodes when he was four years old, which is one of the rarer clinical complications of CPSS and has been described mostly in newborns, in whom it can be clinically very severe and persistent and is most likely due to hyperinsulinism from reduced hepatic degradation of insulin[28]. In our case, the patient responded well to increased oral glucose intake.
Additionally, with CPSS there is an increased incidence of benign and malignant hepatic lesions[2,7,29,30]which are presumed to be linked to decreased perfusion of hepatic tissue with portal blood and concomitantly increased hepatic arterial blood flow[12,13]. Benign liver tumors were present in two patients with extrahepatic CPSS from our series. In one of them, the CPSS was found incidentally during diagnostic work-up for tumor formation in the right liver lobe, which was performed due to increased liver enzymes. The other patient with neonatally-diagnosed CPSS (case 4) had had increased alpha-fetoprotein levels since the newborn period and had multiple tumor formations, observed on US when he was four years old. Since these changes were stable and had the US appearance of regenerative nodules, no invasive treatment has been necessary up to now. Both patients continue to receive serial imaging investigations in order to detect potential malignant alteration.
We also present two unusual complications of CPSS, upper gastrointestinal bleeding and osteoporosis.
The first patient presented with severe, recurrent gastrointestinal bleeding, which is a rare clinical manifestation of CPSS. Gastrointestinal bleeding in CPSS was previously reported in 2015 by Gonget al[31], who described six patients that initially presented with bleeding from the lower gastrointestinal tract and were all found to have superior rectal vein and colonic varices due to an extrahepatic shunt that drained portal blood into the iliac veinviathe inferior mesenteric vein. This was also reported to be the most common type of CPSS associated with gastrointestinal bleeding by Kobayashiet al[32], who reviewed clinical features of 136 published cases of extrahepatic CPSS, 8% of which were associated with gastrointestinal bleeding. In contrast to the most commonly reported association of gastrointestinal bleeding with extrahepatic CPSS, the patient we describe had an intrahepatic CPSS between the left portal and left hepatic vein. He presented with melena, which indicates that the bleeding was of upper gastrointestinal origin and not due to rectal or colonic varices, as described by Gonget al[31]Of the previously published cases of CPSS with gastrointestinal bleeding, only one patient reported by Alomariet al[33]had an intrahepatic CPSS, as our patient did. They proposed that the bleeding was caused by diversion of the mesenteric blood flow from the bowel, which resulted in relative intestinal mucosal ischemia, with diffuse erosive changes in the intestine[33]. In our patient, prominent gastric submucosal vasculature (consistent with hypertensive gastropathy) was seen two years after his first presentation, when the abdominal CT with contrast also revealed an enlarged venous plexus at the gastric antrum. This finding is somewhat unusual, however, since portal hypertension normally does not develop in CPSS, because the blood flows easily through the shunt[6].
Osteoporosis was present in two of our patients: In a 14-year-old girl, severe osteoporosis was the first clinical sign of CPSS, and a boy was discovered to have osteoporosis 4 years after he was diagnosed with CPSS (case 4). Cholestatic liver disease is known to be frequently accompanied by hepatic osteodystrophy but, to the best of our knowledge, the correlation of osteoporosis and CPSS has not previously been studied. Severe osteoporosis was reported in a 17-year-old patient with extrahepatic CPSS and several others concomitant anomalies in a case series by Ponzianiet al[11], but this association has not been explored further. Studies on hepatic osteodystrophy postulate, however, that, in addition to hepatic dysfunction, portal blood flow abnormalities and malabsorption could contribute to its pathogenesis. van der Merweet al[34]showed that portosystemic shunting is a major pathogenic factor causing bone loss in rats, but this pathophysiologic mechanism has not yet been studied in humans. Abnormalities in vitamin D metabolism or vitamin D deficiency may also play a role in the development of osteoporosis in CPSS. The girl with severe osteoporosis (case 5) was found to have 25-OH vitamin D, on the lower limit of normal and in the boy from case 4, osteoporosis developed despite vitamin D3 supplementation. Other patients from our series also had vitamin D, as well as other fat-soluble vitamins deficiencies and are receiving regular replacement therapy. This underscores the importance of checking the levels of fat soluble vitamins in patients with CPSS. No clinical reports have highlighted this issue thus far, with the exception of coagulopathy reflected in prolonged prothrombin time, which was described in 31 of 77 patients reviewed by Bernardet al[2]and in 2 of 3 patients from a case series by Fuet al[35]. Among our patients, coagulopathy responsive to vitamin K substitution was detected in three children. A possible mechanism for fat soluble vitamin deficiencies in CPSS could be a disruption in the enterohepatic circulation of bile acids as a result of intestinal blood bypassing the liver. Anoxic-ischemic cholestasis due to liver underperfusion could also be a contributing factor.
When it comes to treatment, occlusion of type 1 CPSS is not possible in most cases, because the shunt is the only possible drainage pathway for blood from the mesenteric and splenic veins[2,36], which is best proven by portography after occlusion of the shunt. The only treatment option for patients with no intrahepatic portal venous system who develop severe complications is therefore liver transplant, which was performed in the patient with severe porto-pulmonary hypertension from our case series, in whom a total absence of the intrahepatic portal vein was confirmed with wedge venography. The two other patients with extrahepatic CPSS are clinically stable and are not yet on the active liver transplant list. This is in accordance with the reports from Sokolliket al[7]and Knirschet al[16], who described several patients in good clinical condition on symptomatic treatment. It is important to note, however, that we did not perform portography in these two patients and therefore the correct malformation subtype might not have been identified.
After confirming that the intrahepatic portal system is intact, partial CPSS can be occluded either by surgical ligation or percutaneously by an interventional radiologist[2,16,18], such as was successfully performed in our first case, in whom the biopsy showed normal hepatic tissue with venules present in the portal triads
Stabilization and regression of the pulmonary, neurologic, cardiac and vascular complications can be expected after liver transplantation or shunt resolution[2,15,35-37]. Jainet al[9], for example reported marked improvement in clinical signs, symptoms and laboratory values in all five patients with extrahepatic CPSS type 2 who underwent shunt ligation and complete closure of the shunt stabilized or even reduced pulmonary arterial pressure in two of the patients from a case series by Kirschet al[16]. Orthotopic liver transplantation has similarly good results, as reported by Xianget al[36]. In our series, we discovered an improvement of hypertensive gastropathy in the first patient from our case series. In the patient who underwent liver transplant, pulmonary hypertension did not improve significantly at least in long term.
In the second child of our patients, who had an intrahepatic shunt discovered in the neonatal period, spontaneous regression was observed and, according to the published literature, the shunt will probably close with time[2,7,17,18].
The principle limitations of our study are the relatively small sample size due to the rarity of the condition and the size of our centre, and retrospective data analysis with data quality depending on the accuracy of the medical records and the extent of diagnostic workup. Because patients were diagnosed at different time points during the 15-year period, the length of follow-up and the age at the end of the study were variable. Another possible limitation is the aforementioned lack of imaging of the portal venous system by portography which may have led to diagnostic error in some of our patients.
CONCLUSION
In conclusion, Abernethy syndrome is a rare congenital anomaly that can affect all organ systems and most commonly presents with neonatal cholestasis, hyperammonemia, pulmonary arterial hypertension and liver tumors. Rarer clinical manifestations of CPSS are also possible, such as upper gastrointestinal tract bleedings and bone fractures. Along with the well-known correlation of CPSS with congenital cardiac disease, we also describe a possible association with other congenital vascular anomalies, such as Klippel–Trénaunay syndrome, which should perhaps prompt screening for CPSS. Our patients were treated according to the existing algorithms, based on the anatomy of the lesion and the clinical picture itself and continue to receive close follow-up.
 World Journal of Gastroenterology2020年37期
World Journal of Gastroenterology2020年37期
- World Journal of Gastroenterology的其它文章
- Artificial intelligence technologies for the detection of colorectal lesions: The future is now
- Transjugular intrahepatic portosystemic shunt in cirrhosis: An exhaustive critical update
- Endoscopic retrograde cholangiopancreatography in the treatment of pancreaticopleural fistula in children
- Risk prediction rule for advanced neoplasia on screening colonoscopy for average-risk individuals
- Endoscopic ultrasound-fine needle biopsies of pancreatic lesions: Prospective study of histology quality using Franseen needle
- Helicobacter pylori infection with atrophic gastritis: An independent risk factor for colorectal adenomas