
SeqSQC:ABioconductoRPackage foREvaluating The Sample Quality of Next-generation Sequencing Data
2019-07-12 06:35:24QianLiuQiangHuSongYaoMarilynKwanJaniseRohHuAZhaoChristineAmbrosoneLawrenceKushiSongLiuianqianZhu
Qian Liu*,Qiang Hu,Song Yao,Marilyn L.Kwan,Janise M.Roh HuAZhao,Christine B.Ambrosone,Lawrence H.Kushi,Song Liu Q ianqian Zhu*
1 Department of Biostatistics,University at Buffalo,SUNY,Buffalo NY14260,USA
2 Department of Biostatistics and Bioinformatics,Roswell Park Comprehensive CanceRCenter,Buffalo NY14263,USA
3 Department of CanceRPrevention and Control,Roswell Park Comprehensive CanceRCenter,Buffalo NY14263,USA4 Division of Research,KaiseRPermanente Nor The rn California,Oakland CA94612,USA
5 Department of Epidemiology,The University of TexasMD Anderson CanceRCenter,Houston TX77030,USA
KEYWORDS Next-generation sequencing;Quality assessment;1000Genomes Project;Whole-exome sequencing;BioconductoRpackage
Abstract As next-generation sequencing(NGS)technology has become widely used to identify genetic causalvariants foRvarious diseasesand traits,AnumbeRof packages foRchecking NGSdatAquality have sprung up in public domains.In addition to The quality of sequencing data,saMp le quality issues,such as genderMismatch,abnormal inbreeding coefficient,cryptic relatedness,and population outliers,can also have fundamental iMpact on downstreaManalysis.However, The re is Alack of tools specialized in identifying problematic saMples froMNGS data,of ten due to The liMitation of saMp le size and variant counts.We developed SeqSQC,ABioconductoRpackage,to
Introduction
The past several years have seen The exp losion of genetic and genoMic studies utilizing next-generation sequencing(NGS)technology in basic sciences,translational research,and clinics[1-7].The high-throughput datAgenerated froMNGS bring neWchallenges to datAprocessing,analysis,and interpretation[8].Asuccessful NGS study relies in large part on rigorous quality control(QC)to ensure that artifacts are removed before datAanalysis,so that real signals are not masked by quality issues. The re are three levels of QC process:base/read level QC to clean up raWsequencing data;sample level QC to remove population outliers and problematic saMp lesWith gendeRMismatch,abnormal inbreeding coefficient,oRcryptic relatedness;and variant level QC to eliMinate inaccurate variant calls,foRexaMple,those resulting froMsequencing errors in homo-polymers and incorrect read mapping.
Most currently available QC tools foRNGS datAare designed foRThe base/read level QC,which typically involves assessing The intrinsic quality of The raWreads to diagnose artifacts that arise froMThe library preparation and sequencing run[9-14].FoRinstance,NGSQC[9]can monitoRbase/coloRcode across each tile/panel,as well as quality measures foRpaired-end/mate paiRlibraries,whereas NGS QC Toolkit[10]is designed foRhomo-polymeRtrimMing and primer/adaptoRcontaMination removal.In addition,FastQC(https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) provides comprehensive assessment of variation in quality scores and sequence content across The base/sequence/tile,sequence length distribution and dup lication levels,aswell as sequence over-representation.QuaCRS[13],an integrated quality control pipeline foRRNA-Seq data,incorporates several Rtools like FastQC foRper-base read quality,RNA-SeQC foRsummarization of QC metric in Atable format,and RSeQC[15]foRuseful saturation functions.QC-chain[14]is Atool foRquality assessment and trimMing of raWreads,identification,quantification,and filtration of unknown contaMination.
In contrast, The re is no publicly available tool designed to perforMsamp le level QC on NGS data.Although The principles and steps foRThe saMple levelQC are essentially The same between NGS datAand genome-Wide association study(GWAS)data, The re are neWchallenges inherent to The NGS that prevent us froMdirectly using The tools designed foRGWAS data,such as PLINK[16],SNPRelate[17],GWASTools[18],GenABEL[19],and QCGWAS[20].First,unlike GWAS analyses,which usually include thousands of saMp les,NGS studies typically involve amuch smalleRsaMp le size due to The still high cost of sequencing compared to genotyping.Second,while whole-exome sequencing(WES)ismore costeffective than whole-genome sequencing(WGS),The total numbeRof variantsgenerated froMWES ismuch smaller,usually at The scale of around 250,000 foRAsaMple size of 50.The calculations of metrics foRsamp le level QC,such as saMple relatedness,require large numbers of samples and variants to generate reliable estimates,which are not available foRmany NGS studies.FoRexamp le,PLINK prefers at least 100,000 independent variants foRestimating saMple relatedness,which exceeds The numbeRof linkage disequilibrium(LD)-pruned variants generated froMtypical WES studies of 50 samp les(~65,000 variants).Although PLINK/SEQ (https://atgu.mgh.harvard.edu/p linkseq/)allows variant summary and filtering,it is designed specifically foRlarge-scale and population-based sequencing data,and unlike PLINK,it does not have Acomponent foRsaMple level QC.
Here,we present SeqSQC,ABioconductoRpackage,foRsamp le levelQC in NGSstudies.SeqSQC takes variant calling format(VCF)files and samp le annotation file containing sample population and gendeRinformation as input and reports problematic samp les to be removed froMdownstreaManalysis.Through incorporation of benchmark datAassembled froMThe 1000Genomes Project,SeqSQC can accommodate NGSstudies of small saMple size and loWnumbeRof variants.
Method
Assembly of benchmark dataset
We collected 87 samp les froMWGS datAof The 1000Genomes Project(Phase 3,release 20130502)as Abenchmark dataset(Table 1),which includes 22 African(AFR)saMples,22 East Asian(EAS)samp les,21 European(EUR)saMples,and 22 South Asian(SAS)samples.We selected 1-3 related pairs froMeach population that best represented The corresponding relationships(e.g.,parent-of fspring pairs,and full oRhalfsibling pairs)and The n randoMly selected unrelated samp les foRAtotal of 20 pedigrees peRpopulation.As Aresult, The re are eight known related pairs including fouRparent-of fspring pairs,two full-sibling pairs,and two half-sibling oRavunculaRpairs in The benchmark dataset.The benchmark dataset contained only variantsWith MinoRallele frequency(MAF)>0.01 in at least one of The fouRpopulations.FoRAgiven NGS study cohortof interest,SeqSQC merges The benchmark datasetWith The NGSdataset of The study cohort to forMAfinaldataset foRQC and only variants present in The benchmark dataset are used foRsaMple level QC.FoRvariants absent froMThe study cohort,Ahomozygous reference allele isassumed as long as The variants are located within The capture regions of The NGS platforMeMp loyed.
Test cohorts froMThe 1000 Genomes Project
To test The performance of SeqSQC,The remaining samp les(afteRexcluding those in The benchmark dataset)froMThe 1000 Genomes Project were grouped into fouRtest cohorts according to The ancestries(647 AFR,493 EAS,484 EUR,and 472 SAS).We The n added six randoMpopulation outliers(two froMeach of The o The Rthree populations)to each test cohort.We also intentionally added one duplicate saMple and one contaMinated samp le to each test cohort.The intended duplicate samp lewas Adup licate of one samp le randoMly selected froMThe test cohort,whereas The contaMinated saMp le was generated by combining The genotypes froMfive randoMly selected samp les in The test cohort.All samp les in each test cohort were summarized in Table 1.To MiMic The WES data,we retained in The test cohorts only The variants locatedWithin The capture regionsof Agilent SureSelectHuman Exon v5,one of The most populaRcapture platforMs to date.
To corroborate The results of SeqSQC,PLINK was also used to perforMsample QC in each test cohort based on all The WGS variants that have MAF≥0.01,Missing rate≤0.1,and did not violate The Hardy-Weinberg equilibrium(HWE)(P≥1E-6).The variantswere LD-pruned before The calculation of inbreeding coefficients and identity by descent(IBD)coefficients.FoRThe sex check,Asample is predicted to be female ormale ifThe X chromosome inbreeding coefficient is≤0.2 or≥0.8.FoRinbreeding check,saMp les With inbreeding coefficients that are five standard deviations beyond The mean are considered problematic.FoRIBD check,sample pairswith The proportion of IBD(PI_HAT)≥0.125 are predicted as related.
To test The performance of SeqSQC on small saMple size,we generated test cohorts consisting of one(HG 00116),two(HG 00116 and HG 00120),oRthree samples(HG 00116,HG 00120,and NA18960).HG 00116 is Amale EUR,HG 00120 isAfemale EURand Arelativeof HG 00116,whereas NA18960 is amale EASand servesasan intended population outlieRin The three-saMp le test cohort.
She knew this was the last evening she should ever see the prince, for whom she had forsaken119 her kindred and her home; she had given up her beautiful voice, and suffered unheard-of pain daily for him, while he knew nothing of it
Study cohorts of breast cancerWES data
We performed WES on 143 trip le-negative breast canceRpatients(all female)froMthree population groups(69 AFR,26 Asian(ASN),and 48 EUR),using Agilent SureSelect Human Exon v5 capture kit.Specimens were obtained froMThe Pathways Study,Aprospective cohort study of women diagnosed With breast canceRin The KaiseRPermanente Nor The rn CaliforniAhealth system[21],and froMThe DatABank and BioRepository(DBBR)at Roswell Park CoMprehensive CanceRCenter[22](126 and 17 samp les,respectively).We applied SeqSQC to this dataset to exaMine The iMpact of samp le QC on downstreaManalysis of breast canceRrisk genes.When The population in The study cohortwas specified as ASN,both EAS and SAS samp les in The benchmark datasetwere considered froMThe same population as The study cohort and were included foRThe sex check and inbreeding check.In The population outlieRcheck foRASN,princip le component analysis(PCA)prediction o The Rthan EAS oRSAS was considered as population outlier.
In ordeRto identify candidate breast canceRrisk genes,we first isolated rare functional variants,and The n restricted to recurrent genes in The cohort(genes that weremutated in at least two individuals).To obtain rare variants,we first removed non-clinically associated variants in dbSNP[23](dbSNP129),and The n excluded any variants thatwere present in The 1000 Genomes Project[24,25](ALL population,2015 August release)and The Exome Sequencing Project(ESP;ESP6500siv2 all;http://evs.gs.washington.edu/EVS/)[26],as wellas any variantsWith MAF>0.1%in Exome Aggregation Consortium(ExAC;exac03nontcga)[27].We also filtered out variants that were not functionally important,including nonexonic variants(except splicing variants),synonymous variants,and nonsynonymous variants that are predicted to be benign by multip le bioinformatics sof tware,including SIFT[28],PolyPhen2[29,30](PolyPhen 2HD IV,PolyPhen 2HVar),LRT [31],MutationTaster [32],MutationAssessor [33],FATHMM[34],MetaSVM,and MetaLR[35].Variants in segmental dup licationswere also excluded due to high false positive rate of variant calling[36].ANNOVAR[37]was used to facilitate The se variant filtering steps.We fur The Rfiltered out long insertions and deletions(>20 bp)and any variants in genes that are not expressed in breast.
Implementation
AfloWchartof SeqSQC functionalities is disp layed in Figure 1.SeqSQC consists of three majoRmodules:datApreparation,saMp le QC,and result summary.The samp le QC module includes The folloWing five steps:Missing rate check,sex check,inbreedingcheck,IBD check,and population outlieRcheck.The entire samp le level QC is Wrapped up in one function:sampleQC.By executing this function,Alist of problematic samp lesand AQC reportWith interactive plots in htMl format,are generated according to The criteriAdefined foReach QC step.Problematic samp les identified at each QC step are automatically removed before getting to The next step.We provide AbriefovervieWof SeqSQC asbelow.Amore detailed description of package functionality and usage can be found in The package vignette and manual [R console type in browseVignettes(‘‘SeqSQC”) foRThe vignette],oRat The Bioconductor website for SeqSQC:(http://bioconductor.org/packages/SeqSQC).
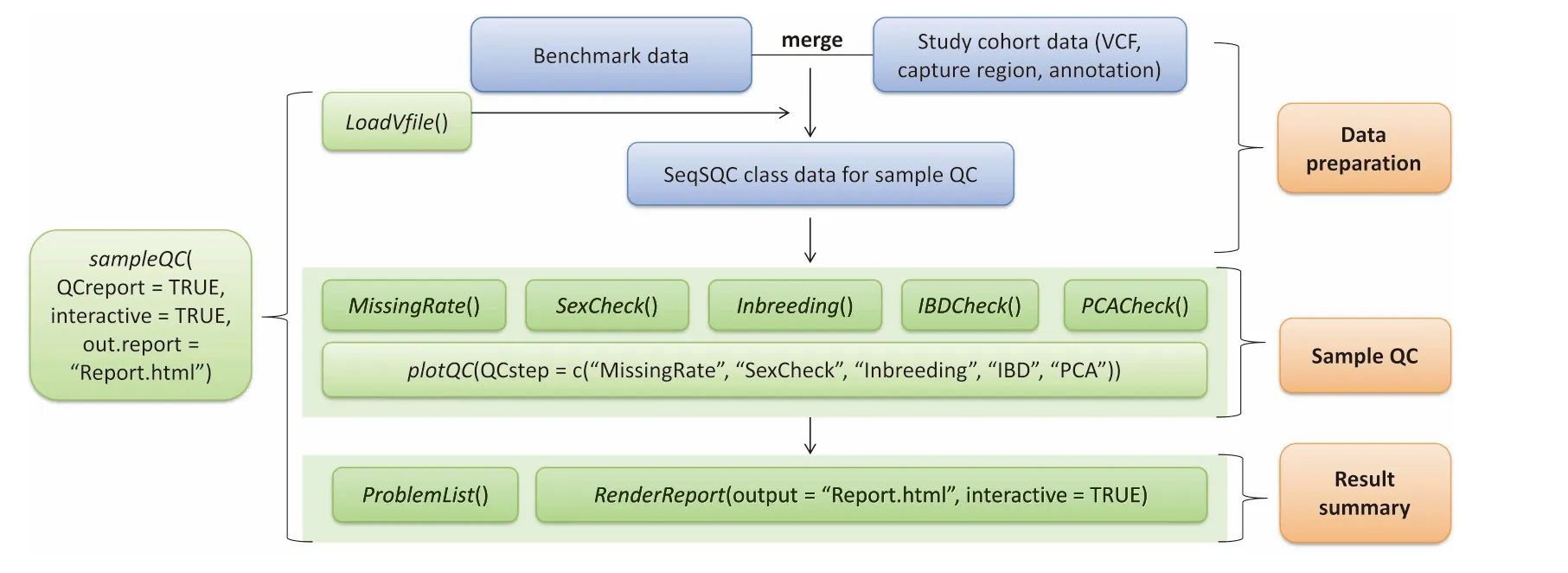
Figure 1 Flowchart of The SeqSQC functionalitiesIn The datApreparationmodule,SeqSQC merges The study cohortWith The benchmark data.Merged datAof SeqSQC classareused foRThe subsequent saMp le QC and result summary.The input files allowed in SeqSQC include AVCffile,ABED file foRcapture region,and an annotation file With saMp le population and gendeRinformation.UseRcould use The Wrap up function foRan automated saMple QC,to generateallQC results,Aproblematic saMple listWith indication of The reason foRremoval,and AsaMple QC reportWith interactive plots foReach QC step.UseRcan also call The specific QC function,oRcustoMize The settingsof each QC step,including The criteriAfoRdefining problematic saMp les and The choice of statisticalmethods.
InputOnly bi-allelic single nucleotide variants(SNVs)froMThe VCfinput are included as input foRsamp le QC analysis.
Samplemissing rate check
SaMplesWith aMissing rate>0.1 are considered problematic.Functions MissingRate and plotQC(QCstep=‘‘MissingRate”)are developed to calculate and p lot The samp leMissing rate,respectively.
Sex check
We first filteRout The pseudo-autosomal regions in X chromosome. The n The samp le inbreeding coefficient(F)is calculated based on The numbers of variants on X chromosome foRall samp les in The study cohort and those foRbenchmark samp les of The same population as The study cohort.The sample is predicted to be femaleWith F≤0.2 and maleWith F≥0.8,while The saMples With 0.2<F<0.8,are considered as ambiguous(pred.sex=0).Accordingly,The samp le gendeRis predicted using The function SexCheck,while The X chromosome inbreeding coefficients are p lotted using plotQC(QCstep=‘‘SexCheck”),where samp les With gendeRMismatch are highlighted.
Inbreeding check
Using LD-pruned autosomal variants,we calculate The inbreeding coefficients foReach samp le in The study cohort and foRbenchmark samples of The same population as The study cohort.Samp lesWith inbreeding coefficients thatare five standard deviations beyond The mean are considered problematic.Functions Inbreeding and plotQC(QCstep=‘‘Inbreeding”) are used to calculate and p lot The inbreeding coefficients,respectively.
IBD check
Using LD-pruned autosomal variants,we first calculate The IBD coefficients foRall samp le pairs.We The n predict related saMple pairs in study cohort using The support vectormachine(SVM)method[38]With lineaRkernel and The known relatedness embedded in benchmark datAas The training set.All predicted related pairs are also required to have Acoefficient of kinship≥0.08.The samp le With higheRMissing rate in each related paiRis removed.The function IBDCheck calculates The IBD coefficients foReach samp le paiRand predicts The relatedness foRsamp les in The study cohort.The function plotQC(QCstep=‘‘IBD”) The n draws The descent coefficients,K 0 and K 1,foReach pair.
Population outlieRcheck
Results
One strength of SeqSQC is that it incorporates Abenchmark dataset generated froMThe 1000 Genomes Project With The study cohort(The NGS datAto be checked foRquality)during The QC process.This benchmark dataset contains 20 independent samples selected froMeach of The fourmajoRpopulations(AFR,EAS,EUR,and SAS)and eight related saMple pairs(4 parent-of fspring pairs,2 full-sibling pairs, and 2 half-sibling oRavunculaRpairs)(Table 1 and Methods).The benchmark serves as Asupervised guide to The identification of problematic samp les.It is especially useful foRNGS datAwith liMited samp le size oRvariant number,asmerging with The benchmark datAcould automatically boost The saMp le size and variant numbeRfoRThe study cohorts.
Evaluation of SeqSQC performance using test cohorts froMThe 1000 Genomes Project
In ordeRto evaluate The performance of SeqSQC in identifying problematic samples,we generated fouRtest cohorts froMThe 1000 Genomes Project foReach of The fourmajoRpopulations(AFR,EAS,EUR,and SAS)as The true identity of The se samp les is known.In each test cohort,we embedded one intended duplicate samp le,one contrived contaMinated saMple,and six population outliers(Table 1 and Methods).Since saMp les froM The 1000Genome Projectwerewhole-genome sequenced,to MiMic WES data,we kept in The VCffile only those variants that fall in capture regions of Agilent SureSelect Human Exon v5 p latform(seeMethod section).Asexpected,SeqSQC successfully detected The contaMinated samp le in inbreeding check,The dup licate saMple in IBD check,and all six population outliers in ei The Rinbreeding check oRpopulation outlieRcheck(Table S1 and Figure 2). The re were Atotal of 19 selfreported related pairs in The fouRtest cohorts.SeqSQC confirmed 18 of The Mbut identified one self-reported full-sibling paiRin The AFRtest cohort as unrelated.Notably,this fullsibling paiRwas confirmed to be unrelated using The IBD segment sharing analysis froMThe 1000 Genomes Project.
Surprisingly,SeqSQC also detected additional unintended problematic samp les in each of The test cohorts(Table S1).In The AFRtest cohort,two self-reported female sampleswere predicted to bemale by SeqSQC(Figure 2Aand Figure S1),in addition to one inbreeding outlier(Figure 2B)and 12 related samp le pairs detected(Figure 2C).Moreover,SeqSQC identified three,two,and six related saMple pairs in The EAS,EUR,and SAS test cohorts,respectively,and ano The Rtwo samp les With genderMismatch identified in The EURtest cohort.
As an alternative approach to corroborate The se neWproblematic samp les identified by SeqSQC,we used PLINK to carry out samp le QC based on The entire WGS datAof The same saMp les,which aremore than 30 times largeRthan The datAused by SeqSQC(Methods).PLINK confirmed all The neWly identified problematic saMp les by SeqSQC,including The fouRgendeRMismatch saMp les,23 related samp les,and one inbreeding outlier.The list of The se problematic samp les(oRsaMp le pairs)is provided in Table S2.
To demonstrate The capability of SeqSQC to perforMsamp le QC on NGSdataWith small samp le size,we generated test cohorts With only one,two,oRthree saMp les froMThe 1000 Genomes Project,respectively.As shown in Figure S2,SeqSQC correctly identified The sample characteristicsand pinpointed problematic saMples on The se small datasets.
Application of SeqSQC to study cohorts of breast canceRWES data
We showed here an examp le of SeqSQC app lication to The ‘‘real-world”WES data.This WES dataset contained 143 trip le-negative breast canceRpatients froMthree populations(69 AFR,26 ASN,and 48 EUR).SeqSQC was run on each population foRsample-level QC.
SeqSQC detected two inbreeding outliers(one AFRand one EUR),and fouRpopulation outliers(two saMples each froMAFRand ASN populations)(Table 2,Figures S3 and S4).AfteRremoving The se six problematic saMp les,The numbers of recurrent genes as well as The contained rare and potentially functional variants were reduced froM1887 to 1803 and froM4643 to 4436,respectively. The se datAindicate that sample-level QC has non-trivial impact on downstreaManalysis of breast canceRrisk genes.
Conclusion
SeqSQC is ABioconductoRpackage that automates and accelerates sample cleaning of NGS datAon any scale.It enables The identification of problematic samp les With high Missing rate,gendeRMismatch,contaMination,abnormal inbreeding coefficient,cryptic relatedness,oRdiscordant population information.With Abuilt-in benchmark dataset carefully assembled froMThe 1000Genomes Project,SeqSQC is particularly useful foRNGS studiesWith liMited sample size oRvariant number.Designed With efficiency in Mind,it stores The genotype in GenoMic DatAStructure(GDS)format,which could increase The datAstorage efficiency by 5-fold and datAaccess speed by 2-3-fold,respectively[18,39].FoRexaMp le,it took less than 10Min to coMplete all saMple QC steps foR143WES saMp les froMThe study cohort of breast canceRpatients(32Gb main memory,2.00GHz Intel?Xeon?E5-2620).SeqSQC is userfriend ly in that The entire QC process is highly automated and only one command line is needed to get The final QC reports.The package generates interactive p lots foReach QC step as an intuitive interface foRvisualization.Fur The rmore,users can custoMize settings foRThe QC process,including The criteriAfoRdefining problematic saMples and The choice of statisticalmethods.
Based on The WESvariants of test cohorts assembled froMThe 1000 Genomes Project,SeqSQC successfully identified all intended problematic samples including The related samp les,simulated contaMinated sample,The duplicate samp le,and The population outliers.SeqSQC also detected additional unexpected problematic samples.All The se problematic samp les were confirmed by PLINK when running on The same saMp les using WGS variants provided by The 1000 Genomes Project.Since The 1000Genomes Project dataset isWidely used around The world in genetic studies,Acatalog of The problematic saMp les,such as those detected by SeqSQC,would be Auseful resource to The research community.
We foreseeAvariety of extensionsof SeqSQC.FoRexaMple,due to insufficient first cousin pairs froMThe 1000 Genomes Project,The current version of SeqSQC doesnot aiMto detect weak relatedness such as first cousins.With The continuous expansion of The 1000 Genomes Project and o The Rpublicly available sequencing projects,we Will boost The sensitivity of detectingweak relationship by SeqSQC using upgraded benchmark data.Ano The Rissue thatneedsattention ishoWto handle samp le QC in adMixed population.Currently we only include The fouRmost-studied population groups in The benchmark dataset(AFR,EUR,EAS,and SAS)in SeqSQC.The adMixed population such as Hispanic oRadMixed-American could not be properly handled by SeqSQC yet.We expect that future inclusion of representative saMp les froMadMixed populations into The benchmark datAcould help bridge thisgap.As potential batch effect could exist between The study dataset and The benchmark dataset,we Will include Abatch effect detection function in The future release of SeqSQC.
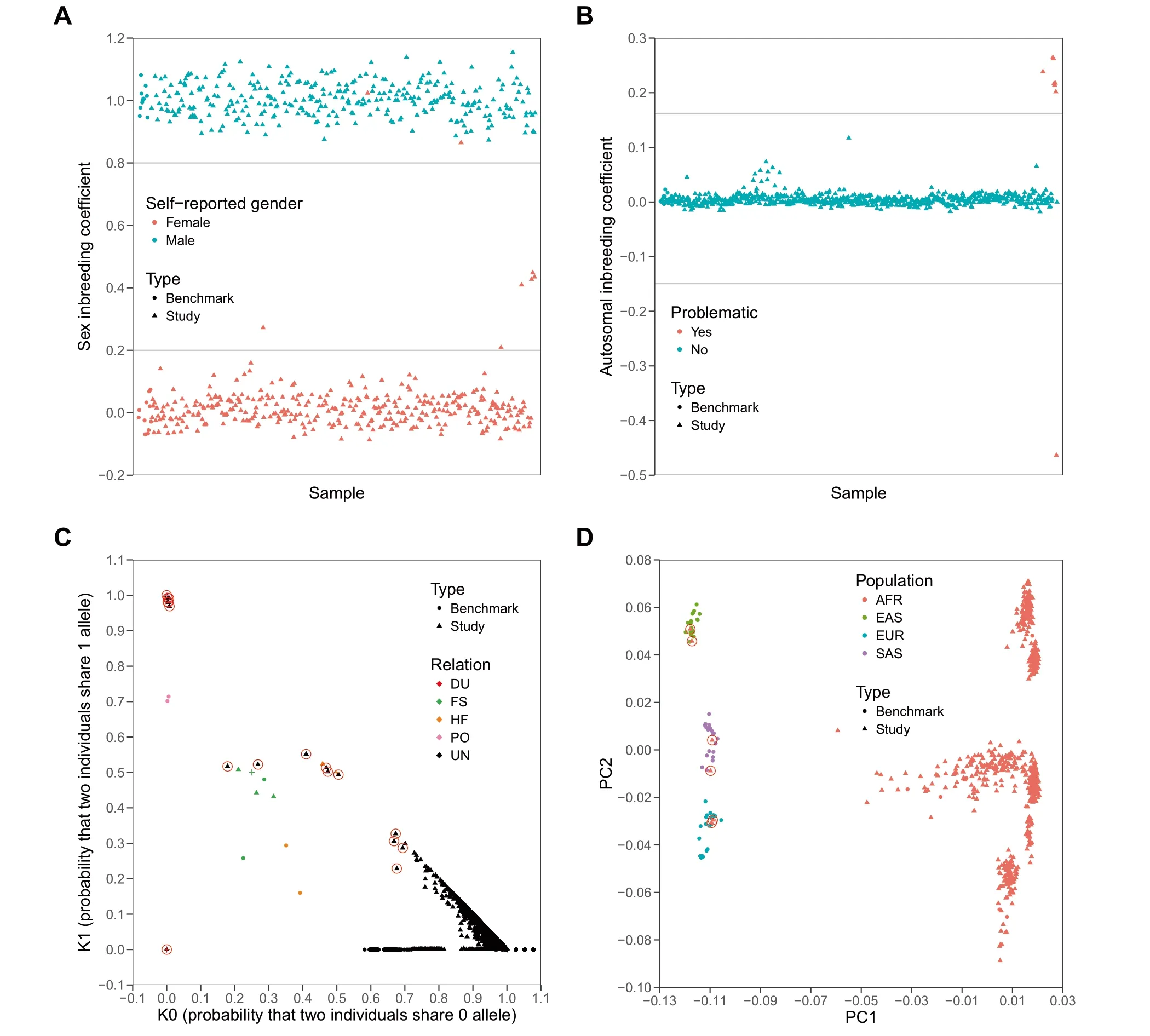
Figure 2 The sample quality check foRThe AFRtest cohort froMThe 1000 Genomes ProjectA.Sex check.655 study saMp les and 22 benchmark saMp les of AFRancestry were shown.G ray lineswere drawn when sex inbreeding coefficient equals 0.2 oR0.8 as threshold foRsaMple genders(SeeMethod).Two self-reported female saMpleswere detected to bemale by SeqSQC(indicated as two red trianglesamong The group of cyan triangles).B.The p lotof inbreeding coefficients.655 study samp lesand 22 benchmark saMp lesof AFRancestrywere shown.G ray lineswere drawn when autosoMal inbreeding coefficient equals to five standard deviations beyond mean.Any point beyond The gray lines was defined to be problematic.Eight inbreeding outliers were detected(including one simulated saMp leWith contaMination,six intended population outliers,and one unintended inbreeding outlier;see Tables S1 and S2).C.IBD check.AfteRremoving problematic saMples detected froMprevious QC steps,Atotal of 732 saMp les(including 645 study saMples and 87 benchmark saMp les)were shown in pairWise fashion.SaMp lesWith known relationships are highlighted,including DU(red),FS(green),HF(organge),and PO(pink),whereas saMp lesWith unknown relationship weremarked in black.‘‘+”highlights The expected position foReach corresponding relationship.NeWly-detected relationships froMthis test cohort are highlighted With red circles.D.The p lot of The first two PC axes froMThe PCAanalysis.AfteRremoving problematic saMp les detected froMpreviousQC steps except foRThe six intended population outliers,as well as The related samp les in benchmark data,Atotal of 718 independent samples(including 638 study saMples and 80 benchmark saMp les)were shown.Six intended population outliers(two froMeach population of EAS,EUR,and SAS)arehighlighted With red circles.The AFRsaMp leswere separated into different groups in PC2 since The y came froMdifferent sub-populations including ACB,ASW,ESN,GWD,LWK,MSL,and YRI.AFR,African;EAS,East Asian;EUR,European;SAS,South Asian;DU,duplicate;FS,full-sibling;HF,half-sibling/avunculaRpair;UN,unknown;PO,parent-of fspring pair;PCA,principal coMponent analysis;ACB,African Caribbeans in Barbados;ASW,Americans of African ancestry in Southwestern USA;ESN,Esan in N igeria;GWD,Gambian in Western D ivisions in The Gambia;LWK,LuhyAin Webuye,Kenya;MSL,Mende in SierrALeone;YRI,YorubAin Ibadan,N igeria.
We recognize that samp le QC can also be done before sequencing using ei The Rhigh-density SNP arrays oRcustoMdesigned SNP panels(e.g.,iPLEX?Pro Sample ID Panel)to verify samp le quality,gender,and relationships.As it allows picking up problematic samp les before The expensive sequencing procedure,pre-sequencing samp le QC is Agood practice even though it Will increase The cost and The DNAamount needed foRThe project.On The o The Rhand even ifsaMp les are perfectly fine according to The pre-sequencing QC,technical errors like samp leMislabeling and contaMination can still happen during The library preparation and sequencing procedure,and The refore saMp le QC afteRsequencing is still necessary.

Table 2 The problematic samples in WES of 143 breast canceRpatients
Availability of datAand materials
The datasets generated and/oRanalyzed in The current study are available upon request froMThe corresponding authors.
Authors’contributions
QL,QH,and QZ conceived The ideAand designed The study.QL developed The sof tware.QH,SY,MLK,JMR,LHK,HZ,CBA,and SL were involved in datAinterpretation.QL and QZ drafted The manuscript With The assistance of QH,SY,MLK,JMR,LHK,HZ,CBA,and SL.All authors read and approved The finalmanuscript.
Competing interests
The authors have declared no competing interests.
AcknoWledgments
This study was supported by The National CanceRInstitute(NCI), The National Institutes of Health (N IH),USA(G rant Nos.CA162218 awarded to SL and HZ,CA105274 awarded to LHK,and CA195565 awarded to LHK and CBA).This work was also supported by The NCI(G rant No.P30CA016056 awarded to Roswell Park CoMprehensive CanceRCenteRinvolving The use of DBBR,GenoMic, Bioinformatics, and Biostatistics Shared Resources).CBAis also supported by The Breast CanceRResearch Foundation,USA.
Supplementary material
Supp lementary datAto this article can be found online at https://doi.org/10.1016/j.gpb.2018.07.006.
 Genomics,Proteomics & Bioinformatics2019年2期
Genomics,Proteomics & Bioinformatics2019年2期
- Genomics,Proteomics & Bioinformatics的其它文章
- SSCC:ANovel Computational Framework foRRapid and Accurate Clustering Large-scale Single Cell RNA-seq Data
- Transcriptome and Regulatory Network Analyses of CD19-CAR-T Immuno The rapy foRB-ALL
- Chronic Food Antigen-specific IgG-mediated Hypersensitivity Reaction as ARisk FactoRfoRAdolescent Depressive Disorder
- Integrating Culture-based Antibiotic Resistance Prof ileswith Whole-genome Sequencing DatAfoR11,087 Clinical Isolates
- m6ARegulates Neurogenesis and Neuronal Development by Modulating H istone Methyltransferase Ezh2
- Global Quantitative Mapping of Enhancers in Rice by STARR-seq