
A novel LC–MS/MS assay for methylprednisolone in human plasma and its pharmacokinetic application
2017-01-19 08:30:30SridhrSiddirjuMeryLzrLlPrsnthTdiboyinSirish
Sridhr Siddirju,Mery Lzr Ll Prsnth*,Tdiboyin Sirish
aDepartment of Pharmaceutical Chemistry,Malla Reddy College of Pharmacy,Osmania University, Secunderabad 500 014,India
bDepartment of Pharmacy,Shri Jagdishprasad Jhabarmal Tibrewala University,Jhunjhunu,Rajasthan 333001, India
cDepartment of Pharmaceutical Chemistry,JSS College of Pharmacy,JSS University,Mysuru 570015,India
Short Communication
A novel LC–MS/MS assay for methylprednisolone in human plasma and its pharmacokinetic application
Sridhar Siddirajua,Mercy Lazar Lal Prasanthb,*,Tadiboyina Sirishac
aDepartment of Pharmaceutical Chemistry,Malla Reddy College of Pharmacy,Osmania University, Secunderabad 500 014,India
bDepartment of Pharmacy,Shri Jagdishprasad Jhabarmal Tibrewala University,Jhunjhunu,Rajasthan 333001, India
cDepartment of Pharmaceutical Chemistry,JSS College of Pharmacy,JSS University,Mysuru 570015,India
A R T I C L EI N F O
Article history:
Received 5 February 2015
Received in revised form 21 June 2015
Accepted 29 June 2015
Available online 28 August 2015
Methylprednisolone
Human plasma
Method validation
LC–MS/MS
Pharmacokinetics
Methylprednisolone is a synthetic glucocorticoid.In our report,the authors proposed a sensitive and selective liquid chromatography/tandem mass spectrometry(LC–MS/MS)assay for the determination of methylprednisolone applying budesonide as internal standard.Liquid–liquid extraction(LLE)havingtert–butyl methyl ether(TBME)have been employed to extract methylprednisolone from the plasma samples.Immediately after reconstitution,the samples were chromatographed on a C18column using an isocratic mobile phase composed of 10 mM ammonium formate buffer and acetonitrile(35:65,v/v).A fow rate of 1.00 ml/min was used to elute the analyte form the column.Analysis was carried out with an API–4000 LC–MS/ MS instrument operated in multiple reaction-monitoring(MRM)mode.The linearity has been proven within the concentration range of 10.1–804 ng/ml in plasma samples.The precision(%CV)and accuracy results in fve validation batches across fve concentration levels were well within the acceptance limits.The drug was stable under different conditions.The particular assay has been profciently put on pharmacokinetic study in healthy male subjects.
?2016 Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license(http:// creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
Synthetic glucocorticoids are frequently prescribed for a broad spectrum of diseases such as allergic,infammatory and autoimmune disorders due to their well-known anti-infammatory and immunosuppressive properties[1].Methylprednisolone (Fig.1)is a methylated derivative of prednisolone,belonging to the group of glucocorticoid or corticosteroid drug.The drug has potent anti-infammatory activity[2]and is used to treat conditions such as arthritis,severe allergic reactions,blood disorders,eye conditions and immune system disorders.Itdecreases the immune system’s response to various diseases to reduce symptoms such as swelling,pain,and allergic-type reactions.About 77%of methylprednisolone bound to plasma proteins in humans.The mean elimination half-life ranges is in the range of 1.8 to 5.2 h.
As per the literature,a lot of LC–MS methods are reported for the determination of methylprednisolone in a variety of biological samples like human plasma[3,4],tissue samples[5–7], human urine[8,9],pig fat[10],bovine milk samples[11,12]and rat plasma[13].Of all the methods reported earlier,only two procedures can be compared with the present work.Recently,Hu et al.[3]reported an LC–MS/MS method for the determination of methylprednisolone in human plasma using multi-step solid phase extraction(SPE)technique for the sample preparation.Initially,sample pretreatment was performed by protein precipitation(PP)and then processed by online SPE technique.Another method is reported by Difrancesco et al.[4]for the determination of methylprednisolone along with other glucocorticoids and immunosuppressive drugs.This method involves intricacy like gradient elution,typical mobile phase consisting of two or more buffers with the pH adjustment, requiring more sample volume(>500 μl)and longer chromatographic run time(>5 min)which are not advantageous if intended for routine drug analysis of pharmacokinetic/ bioequivalence studies.The rest of the analytical methods reported so far were not suitable for routine drug analysis.
To study the pharmacokinetics,tolerability and safety of newer formulations,we need to have a suitable analytical assay for its identifcation along with quantifcation.Nowadays,sensitive and rapid analytical techniques such as LC–MS are most widely used in bio-analytical applications.The current paper details a simple,rapid and selective liquid chromatographyelectrospray ionization tandem mass spectrometry(LC–MS/ MS)assay method for the determination of methylprednisolone in human plasma using budesonide(Fig.1),as an internal standard(IS).The projected LC–MS/MS method is developed with the aim to quantify the methylprednisolone concentrations for a pharmacokinetic and bioequivalence studies.The following are the advantages of the proposed method over those reported earlier:(1)Because of the use of less plasma volume (100 μl),the volume of the sample to be collected per time point from an individual during the study is reduced signifcantly. This allows inclusion of additional points.(2)Employing a single step liquid–liquid extraction procedure minimizes the chances of errors,saves considerable time and simplifes the sample preparation procedure.(3)Greater sensitivity is achieved even with low plasma volumes and the method is well suited for pharmacokinetic analysis.(4)The rapid sample turnaround time of 2.8 min makes it an attractive procedure in high-throughput bioanalysis of methylprednisolone in human plasma.(5)The proposed method was successfully applied to quantify the real time plasma sample concentrations obtained from pharmacokinetic study.Additionally,for the frst time,assay reproducibility is demonstrated through incurred sample reanalysis(ISR).
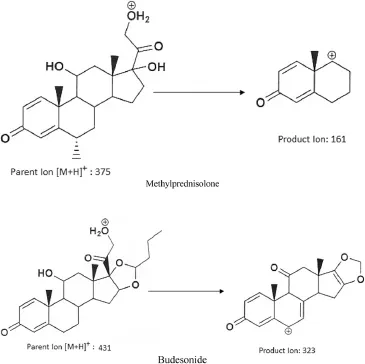
Fig.1–Chemical structures(with fragmentation patch)of methylprednisolone and budesonide(IS).
2.Materials and methods
2.1.Chemicals and reagents
The reference sample of methylprednisolone(98.9%)was purchased from Vivan Life Sciences Limited(Mumbai,India), whereas budesonide was obtained fromAarti Industries Limited (Mumbai,India).HPLC grade methanol and acetonitrile were procured from J.T.Baker(Phillipsburg,NJ,USA).Ammonium acetate was of analytical grade and was obtained from Merck Ltd(Mumbai,India).LC–MS grade water was used for the analysis and was prepared at our laboratory using Milli Q water purifcation(Millipore,Bangalore,India).Six lots of blank plasma containing K2EDTA as an anticoagulant were purchased from Deccan’s Pathological Lab’s(Hyderabad,India).
2.2.LC–MS/MS instrument and conditions
An HPLC system(Shimadzu,Kyoto,Japan)consisting of a Kromasil 100–5 C18,100×4.6 mm,5 μm(Make:AkzoNobel,Pune, India),a binary LC–20AD prominence pump,an auto sampler (SIL-HTc)and a solvent degasser(DGU-20A3)was used for the study.Aliquot of 10 μl of the processed samples were injected into the column,which was kept at(30±2°C).An isocratic mobile phase consisting of a mixture of 10 mM ammonium formate buffer and acetonitrile(35:65,v/v)was used to separate the analyte from the endogenous components and delivered at a fow rate of 1.0 ml/min with splitter(50:50)into the electrospray ionization chamber of the mass spectrometer.Mass spectrometric detection was carried out using AB Sciex API-4000(Foster City,CA,USA)triple quadrupole mass spectrometer equipped with aTurboionspray?in positive ion mode was used for the quantifcation of analyte.The source dependent parameters namely the nebulizer gas(GS1),auxiliary gas(GS2),curtain gas and collision gas were set at 45, 40,20,and 7 psi,respectively.The source voltage was set at5500 V with interface temperature 500°C.The various compound dependent potentials viz.declustering potential(DP), collision energy(CE),collision cell exit potential(CXP)and entrance potential(EP)were set at 97,31,14,10 V for methylprednisolone and 80,20,10,10 V for the IS.Multiplereaction monitoring mode(MRM)was employed for the detection by monitoring the transition pairs ofm/z375.2 precursor ion to them/z161.1 for methylprednisolone andm/z431.3 precursor ion to them/z323.3 product ion for the IS.Quadrupoles resolution was set on unit.Dwell time per transaction was set at 200 ms.Analyst software?(version 1.4.2)was used for the data acquisition.
2.3.Preparation of spiked plasma samples
Methylprednisolone stock solution was prepared in methanol at concentration of 1 mg/ml,whereas further dilutions (working solutions)of methylprednisolone were prepared in a mixture of acetonitrile and water(50:50,v/v;diluent).Two separate stock solutions were prepared and used for the preparation of calibration standards and quality control samples.A 1 mg/ml concentration of budesonide was also prepared in methanol and the working concentration(500 ng/ml)was prepared in the same diluent.
Calibration samples and quality control samples were prepared in K2EDTA human plasma as a bulk.Calibration standards were prepared at nine concentration levels of 10.1, 20.2,40.4,80.8,162,323,482,643 and 804 ng/ml.Similarly,quality control(QC)samples at concentrations of 10.1(lower limit of quantitation quality control,LLOQ QC),30.2(low quality control, LQC),88.9(medium quality control,MQC1),404(MQC2)and 709 ng/ml(high quality control,HQC).All the plasma samples were stored at?70±10°C.
2.4.Sample extraction protocol
All of the frozen plasma samples have been thawed and equilibrated at room temperature just before spiking.An aliquot of 100 μl of human plasma sample was spiked with 20 μl of budesonide dilution(500 ng/ml).After vortexing for 15 s,a 3 ml oftert–butyl methyl ether was added using Dispensette Organic (Brand GmbH,Wertheim,Germany).The sample was shaken for 10 min using a reciprocating shaker and then centrifuged for 4 min at 4200 rpm on Megafuse 3SR(Heraeus,Germany). The clear organic layer(2 ml)was transferred to a 5 ml glass test tube and evaporated at 45°C under a gentle stream of nitrogen.The dried extract was reconstituted with 500 μl of the mobile phase and a 10 μl aliquot of it was injected into the LC–MS/MS system.
2.5.Method validation parameters
A complete method validation of methylprednisolone in human plasma was carried out as per US FDA[14]and EMEA[15]guidelines.The validation parameters determined were carryover test, selectivity,sensitivity,matrix effect,linearity,precision and accuracy,recovery,dilution integrity,long run evaluation, ruggedness and various stability studies.
The selectivity of the method was assessed in six different sources of plasma,of which,four were normal K2EDTA plasma and one each of lipemic and helolyzed plasma.Potential interference from the over-the-counter drugs(paracetamol, nicotine,pantoprazole,ibuprofen,caffeine,diphenhydramine,dicyclomine and pseudoephedrine)were also evaluated. Carryover test ended up being conducted to demonstrate any carryover associated with analyte as well as the IS actuality, which may be revealed throughout following runs.The carryover test samples were injected in the following sequence i.e. blank plasma sample→six samples of LLOQ→blank plasma sample→ULOQ sample→blank plasma samples.Sensitivity was determined by analyzing six replicates of plasma samples spiked with the lowest level of the calibration curve concentrations(LLOQ).Matrix effect,expressed as IS normalized matrix factor(MF)was assessed by comparing the mean area response of post-extraction spiked samples with mean area of aqueous samples(neat samples)prepared in mobile phase solutions at LQC and HQC levels.The overall precision of the matrix factor was expressed as coeffcient of variation(CV).

Matrix effect was also evaluated with six different lots of K2EDTA plasma.Three replicate samples of each of LQC and HQC were prepared from different lots of plasma(36 QC samples in total).The linearity of the proposed method was determined by analysis of fve standard calibration curves(CC) containing nine non-zero concentrations along with one blank plasma sample and one zero standard(blank sample with the IS).Each CC was analyzed individually by least square weighted (1/χ2)linear regression.Intra-day precision and accuracy results were determined using six replicates of LLOQ QC,LQC,MQC–1,MQC–2,and HQC samples analyzed on the same day.Interday precision and accuracy were measured by analyzing fve different analytical batches on three consecutive days.Extraction recoveries of analyte were determined at LQC,MQC–2,and HQC by comparing the peak area of extracted analyte sample with the peak area of non-extracted standard(neat samples). Similarly,recovery of IS was determined at a concentration of 500 ng/ml.Upper concentration(ULOQ)limit was extended by conducting the dilution integrity experiment.Six replicates each at a concentration of about 1.60 times of the uppermost calibration standard were diluted two-and four-fold with blank plasma.The diluted samples were processed and analyzed using un-diluted calibration curve standards.Long run evaluation was carried out to assess the integrity of the samples analyzed in a long run during study sample analysis.A total of 195 samples were analyzed in a single run.The method ruggedness verifed by analyzing one precision and accuracy batch on different column of the same make(different batch no.)using different set of reagents processed by different analyst.
The stock solution stability at room temperature and at 2–8°C in refrigerator was performed by comparing the area response stability samples with the response of the sample prepared from fresh stock solution.The solutions were considered stable if the deviation within±10%form nominal value. Bench top stability at room temperature(15 h),auto-sampler stability for 65 h,wet extract stability for 55 h and reinjectionstability for 37 h,freeze–thaw stability(4 cycles)were performed at LQC and HQC levels using six replicates at each level. Likewise,the long term stability of spiked plasma samples stored at?70±10°C and at?20±5°C for 56 days was also studied at both the QC levels.The stability samples were processed and quantifed against freshly spiked calibration curve standards along with freshly spiked QC samples.Samples were considered to be stable if assay values were within the acceptable limits of accuracy(±15%SD)and precision(≤15%RSD).
2.6.Pharmacokinetic study design
A pharmacokinetic study of methylprednisolone was conducted in healthy South Indian male subjects(n=6).All the volunteers provided written informed consent and the protocol was approved by the local Independent Ethics Committee. A single dose of methylprednisolone tablet(32 mg)was administered orally to all volunteers and blood samples were collected at 0.25,0.5,0.75,1,1.25,1.5,1.75,2,2.25,2.5,2.75,3, 3.25,3.50,4,4.5,5,6,8,10,12,16 and 24 h post-dose.An aliquot of 5 ml blood was collected at each time point in K2 EDTA vacutainer tubes.Additionally,a predose sample was collected to check the possible interferences from the plasma.The collected samples were centrifuged to obtain the plasma and stored at?70±10°C.Plasma samples were spiked with the IS and processed along with QC samples at four concentrations.Pharmacokinetic parameters of methylprednisolone were calculated usingWinNonlin(Version 5.2)software package.Stability of the study samples were established by incurred sample reanalysis(ISR).For ISR two samples from each subject(12 samples in total)were selected nearCmaxand the elimination phase in the pharmacokinetic profle.
The samples were considered stable;the percent difference should not be more than±20%[16,17].
3.Results and discussion
3.1.Method development
To investigate the pharmacokinetics,tolerability and safety of newer formulations,one should have appropriate analytical method for its identifcation and quantifcation.Nowadays,rapid and highly sensitive analytical techniques such as LC–MS are most widely used in bio-analysis of drugs and metabolites.The proposed LC–MS/MS method is developed with the aim to quantify the methylprednisolone concentrations for a pharmacokinetic and bioequivalence studies.Mass parameters were tuned using tuning solution(100 ng/ml)of the analyte.Intensity of the signals obtained in positive mode was much higher than the negative mode.The compound parameters(CE,DP and CXP)were suitably altered to obtain high intense and consist product ion spectra of analyte.Additionally,satisfactory response was obtained by changing the source dependent parameters.A fast and effcient collision cell that can operate at suffcient dwell to enable the acquisition of enough data points across narrow LC peaks with a minimal loss in sensitivity and no cross-talk.Hence,dwell time was set at 200 ms, at which no cross talk was found.Protonated form of analyte and the IS,[M+H]+ion was the precursor ion in the Q1 spectrum and was used as the precursor ion to obtain Q3 product ion spectra.The most sensitive mass transition was observed from m/z 375.2 to 161.1 for analyte(Fig.2A)and from m/z 431.3 to 323.3 for the IS(Fig.2B).Date acquisition was performed with Analyst Software?(version 1.4.2)in the multiple reaction monitoring(MRM)mode.
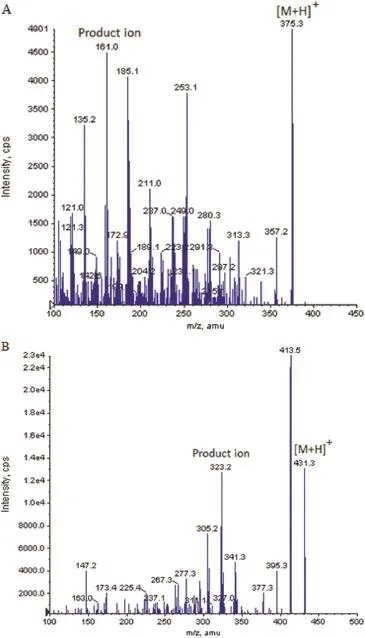
Fig.2–Product ion mass spectra of[M+H]+of(A) methylprednisolone and(B)budesonide(IS).
After selecting the mass spectrometric conditions,chromatographic parameters such as column,mobile phase composition and fow rate were critically assessed to have much better quantifcation results.In general,acetonitrile and methanol are trusted organic modifer intended for LC–MS analysis. Hence,during method development both the solvents were tested in combination with acidic buffers like ammonium formate,ammonium acetate,formic acid and acetic acid.The response obtained with methanol and 5 mM ammonium formate as a mobile phase was suffcient for the quantifcation of the analyte;but the peak shape was not good.Also,avariety of chromatographic columns like C8and C18of different makes(Kromasil 100–5 C18,100×4.6 mm,5 μm;Zorbax SB C18,50×4.6,5 μm;Discovery HS C1850 mm×4.6 mm,5 μm; Alltima HP C1850×4.6,3 μm;Zorbax XDB–phenyl 75×4.6,3.5 μm; Ace 3 C18150×4.6,3 μm;Hypurity advance 75×4.6,5 μm)were verifed to achieve adequate retention time with short run time, better separation from endogenous components,symmetric peak shape and satisfactory response for the analyte.The best chromatographic conditions were achieved with 10 mM ammonium formate and acetonitrile(35:65,v/v)as a mobile phase under isocratic conditions.Kromasil 100–5 C18,100×4.6 mm, 5 μm column gave good peak shape and adequate response even at LLOQ concentration(10.1 ng/ml)level for the analyte. The mobile phase fow rate was set at 1.0 ml/min,which can produce better acceptable chromatographic peak shape and short run time of 2.8 min.
The reported procedures have employed PP[3],SPE[3,5–7,10] and LLE[13]techniques for the extraction of the analyte form plasma.But as the purpose was to develop a simple,rapid and inexpensive method,LLE was tested with ethyl acetate,hexane,tert–butyl methyl ether,dichloromethane and diethyl ether as extraction solvents with/without acidic buffer addition to extract methylprednisolone from the plasma samples.Also,LLE is helpful in producing a spectroscopically clean sample and avoiding the introduction of non-volatile materials onto the column and MS system and also minimizing the experimental cost. Poor recovery results were obtained with the ethyl acetate, hexane,diethyl ether and dichloromethane.Promising results were obtained withtert–butyl methyl ether,which gives clean sample and yields the highest recovery for the analyte and the IS.With or without the addition of extraction additives namely ammonium formate,formic acid and ammonium acetate resulted no difference in the recovery results of the analyte and the IS.Hence,the extraction additives were not added to the plasma samples.
A perfect internal standard should mimic the analyte during ionization,chromatography and extraction.Ideally,a stable labeled isotope drugs are preferably used in LC–MS/MS analysis.But these compounds are expensive and/or not available to serve as internal standard.So,at the initial stages of this work,many compounds were investigated in order to fnd suitable IS;fnally budesonide was selected,based on the chromatographic elution,ionization and extraction effciency.Moreover,the current validation results encouraged its selection as an internal standard.
3.2.Carryover test
Carryover evaluation is most important for desirable precision and accuracy of an analytical procedure.No signifcant carryover was found in blank sample after injection ULOQ sample with the working concentration of the IS indicating that the proposed assay was not compromised due to carryover.
3.3.Selectivity
Selectivity of the proposed method was evaluated by analyzing blank human plasma extract(Fig.3A)and an extract spiked only with the IS(Fig.3B)obtained from six individual sources. As shown in Fig.3A,no signifcant direct interference in the blank plasma traces was observed from endogenous components at the retention time of the analyte and the IS.Similarly, Fig.3B shows the absence of direct interference from the IS to the MRM channel of the analyte.Fig.3C depicts a representative ion-chromatogram for the LLOQ sample(10.1 ng/ml). Similarly,no interference was observed from commonly used drugs such as paracetmol,nicotine,pantoprazole,ibuprofen, caffeine,diphenhydramine,dicyclomine and pseudoephedrine(data not presented).Fig.4 depicts a representative chromatograms resulting from the analysis of subject blank plasma sample along with the IS and 2.25 h subject plasma sample after the single oral dose of a methylprednisolone 32 mg tablet.
3.4.Sensitivity
The lowest measurable concentration with acceptable precision and accuracy for the proposed assay is 10.1 ng/ml,which is set as a lowest limit of reliable quantifcation(LLOQ)for the analyte.At this concentration the signal-to-noise ratio(S/N) was found to be≥10.The precision and accuracy at LLOQ concentration were found to be 3.28%and 107%,respectively.
3.5.Recovery
With the projected LLE method withTBME,good recoveries are obtained for the analyte and the IS.The recovery results obtained for analyte at low,middle and high QC concentrations were 74.9%,75.5%and 74.9%,respectively.The mean overall recovery of methylprednisolone was 75.1±0.36%with the precision range of 1.28–2.07%and the recovery of IS was 73.1% with the precision range of 1.09–1.21%.
3.6.Matrix effect
Matrix effect assessment was done with the aim to check the effect of different lots of plasma on the back calculated value of QCs nominal concentration.The results found were well within the acceptable limits as shown in Table 1.No signifcant matrix effect was observed in all the six batches of human plasma for the analyte at low and high quality control concentrations.Also,matrix factor was calculated at two different concentration levels(LQC and HQC)in each plasma lot and the results are presented in Table 1.
3.7.Linearity,precision and accuracy
The analyte showed good linearity in the concentration range of 10.1–804 ng/ml.A total of fve calibration curves were generated and correlation coeffcient values were in the range of 0.9934–0.9978 for all the analytical runs generated during entire course of validation.The precision and accuracy for the calibration standards ranged from 1.66%to 6.92%and 96.55%to 107.16%,respectively.
The intra-day and inter-day precision and accuracy results in plasma QC samples are summarized in Table 2.The precision(%CV)and accuracy values of methylprednisolone for intraand inter-day ranged 4.19–10.2%and 96.3–105%,and 5.28–9.38%and 100–104%,respectively.The results revealed good precision and accuracy.
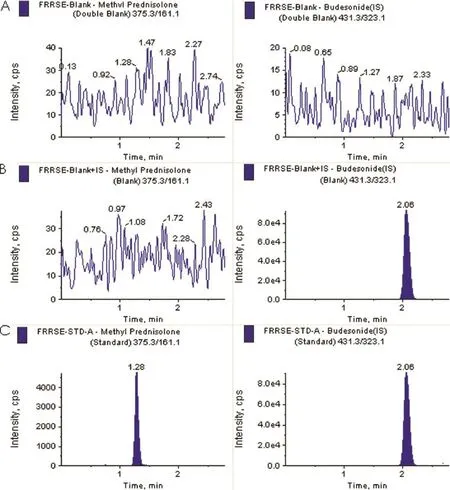
Fig.3–Typical MRM chromatograms of methylprednisolone(left panel)and IS(right panel)in human blank plasma(A), human blank plasma spiked with IS(B)and a LLOQ sample along with IS(C).
3.8.Dilution integrity
The real time subject plasma concentrations above the upper concentration limit(1266 ng/ml)can be quantifed by performing half(1:2)or quarter(1:4)dilution with screened human blank plasma.The precision(%CV)and accuracy values of methylprednisolone for dilution integrity of 1:2 was found to be 1.22%and 94.1%and for dilution integrity of 1:4 dilution was found to be 0.91%and 97.4%.
3.9.Long run evaluation
Long run evaluation experiment was conduct to determine the size of a bio-analytical run during the study sampleanalysis.A set of 195 plasma samples had been processed and analyzed in a single run.160 QCs out of 160 QCs of long run evaluation and 24 QCs out of 24 QCs of freshly spiked were within 15%of their respective nominal(theoretical)values.
The precision(%CV)and accuracy results for long run evaluation QCs were ranged 2.21–2.68%and 98.9–100.2%,respectively. Similarly,the precision(%CV)and accuracy results for freshly spiked QCs were ranged 1.79–3.43%and 97.3–101%,respectively.
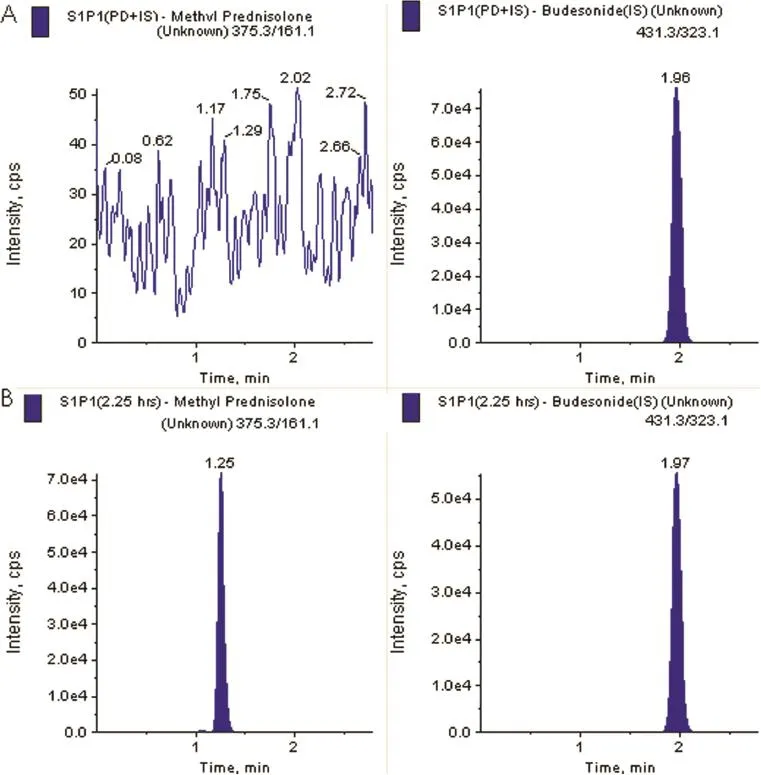
Fig.4–MRM chromatograms resulting from the analysis of subject blank plasma sample(A)and 2.25 h subject plasma sample(B),after the administration of a 32 mg oral single dose of methylprednisolone hydrochloride tablet.The sample concentration was determined to be 312 ng/ml.
3.10.Method ruggedness
Bio-analytical method ruggedness was established with one precision and accuracy(Batch–V)batch.The same samples were processed by the different analyst using different sets of reagents and analyzed on the different instruments of the same make by using different analytical columns(different batch no.). The precision(%CV)and accuracy values for ruggedness batch were ranged 1.79–3.43%and 97.1–101%,respectively.
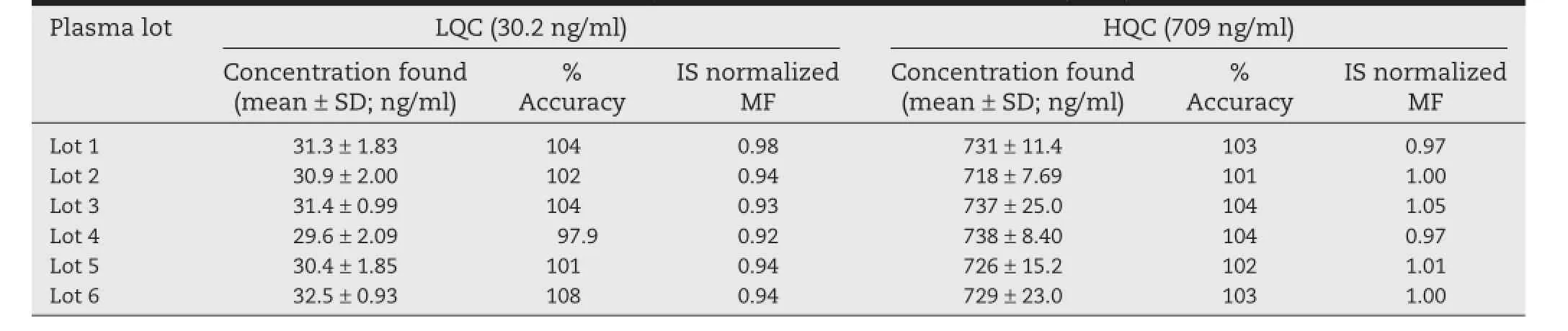
Table 1–Matrix effect assessment results of methylprednisolone in human plasma(n=3).
3.11.Stability studies
A wide range stability experiments namely bench top stability(15 h),repeated freeze–thaw cycles(4 cycles),auto-sampler stability(65 h),reinjection stability(37 h),wet extract stability(55 h at 2–8°C)and long-term stability at?20°C&?70°C for 56 days had been performed throughout validation.The mean%nominal values were found to be within±15%of the predicted concentrations for the analyte at their LQC and HQC levels and the precision(%CV)values were within 15%(Table 3). All the stability study outcomes had been discovered for being inside the specifed limits over the total validation.
3.12.Pharmacokinetic results and incurred samples reanalysis
The proposed assay procedure is successfully applied to a pharmacokinetic study in healthy male South Indian subjects(n=6). The mean concentration(Cmax)in plasma(411±44.2 ng/ml)for methylprednisolone was attained at 2.39±0.74 h(tmax)with ahalf-life(t1/2)of 3.68±0.49 h.The area under the plasma concentration–time curve from time zero to last measurable time point(AUC0–t)and area under the plasma concentration time curve from time zero to infnity time point(AUC0–inf)for methylprednisolone were 2644±464 and 2697±485 ng h/ml, respectively.The mean plasma concentrationvstime profle of methylprednisolone is shown in Fig.5.The study data obtained are authenticated by means of ISR and the obtained results are presented in Table 4.The results demonstrated that the excellent reproducibility of the proposed method.
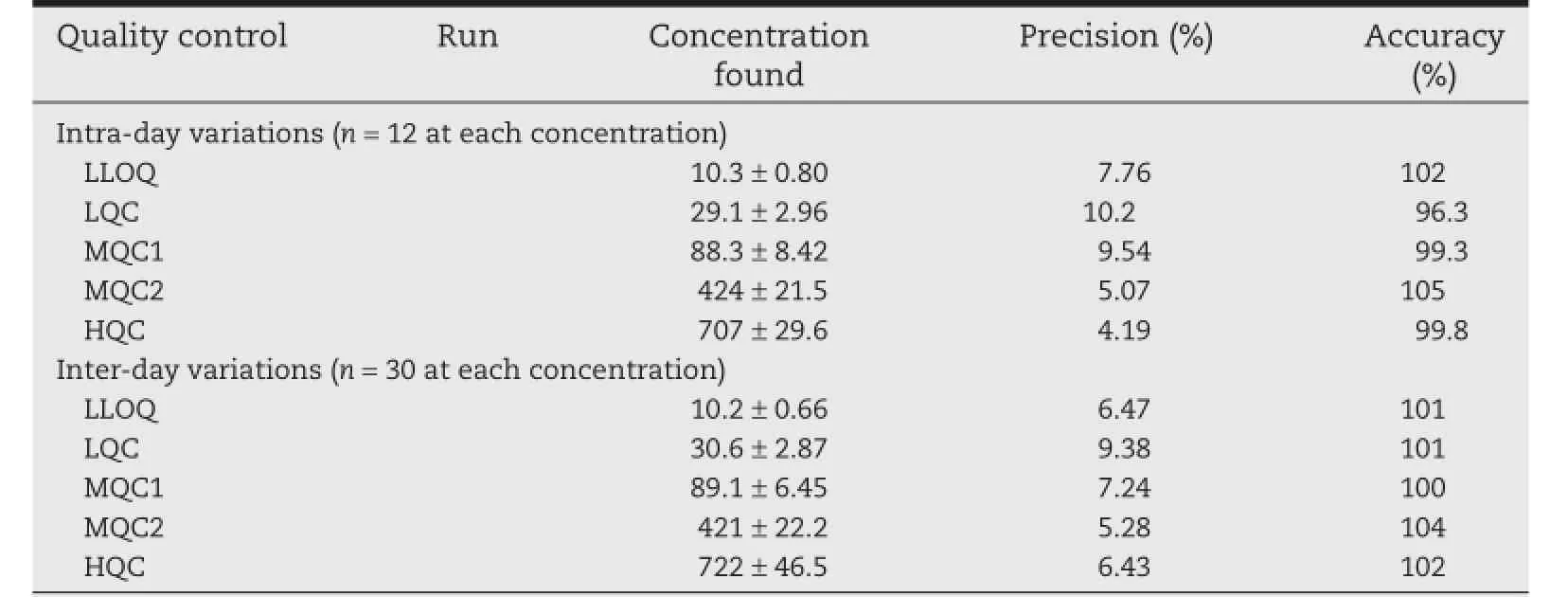
Table 2–Precision and accuracy data for methylprednisolone.
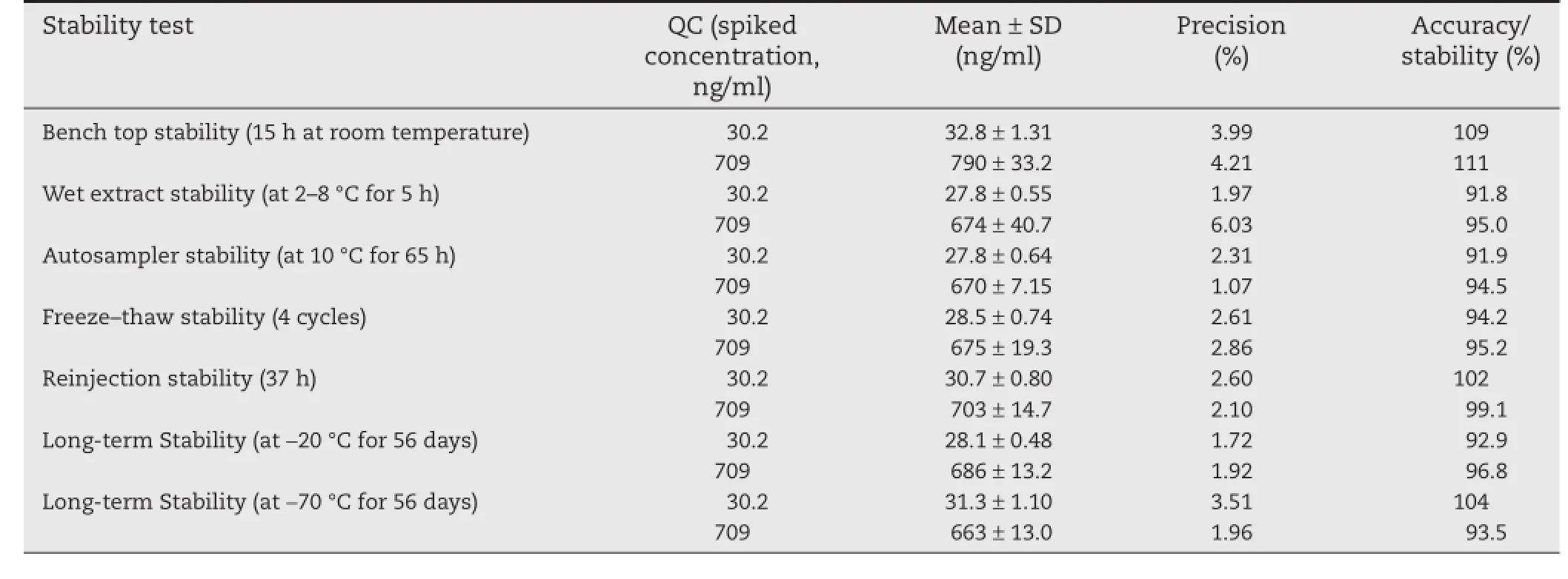
Table 3–Stability data for methylprednisolone in plasma(n=6).
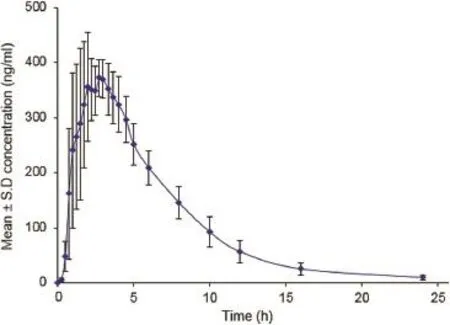
Fig.5–Mean plasma concentration–time profle of methylprednisolone in human plasma following oral administration of methylprednisolone hydrochloride (32 mg tablet)to healthy volunteers(n=6).
4.Conclusions
The LC–MS/MS assay reported here is simple,rapid,specifc and sensitive for quantifcation of methylprednisolone in human plasma and is fully validated according to commonly acceptable US FDA and EMEA guidelines.The simple LLE extraction with TBME extraction method gave consistent and reproducible recoveries for the analyte from the human plasma. The method provided good linearity.The stability of the analyte in plasma and in aqueous samples under different conditions has been extensively evaluated.A sample turnover rate of less than 2.8 min makes it an attractive procedure in highthroughput bioanalysis of methylprednisolone.The method was found to be reliable and reproducible to support pharmacokinetic study in humans.From the results of all the validation parameters,we can conclude that the developed method can be useful for bioavailability and bioequivalence(BA/BE)studies and routine therapeutic drug monitoring with the desired precision and accuracy.
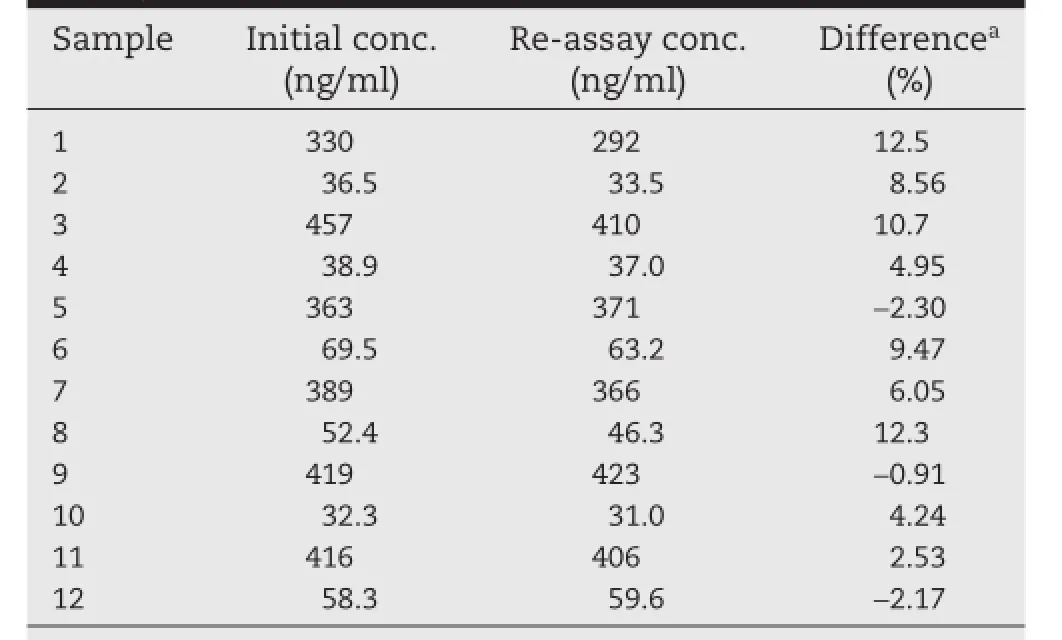
Table 4–Incurred samples re-analysis data of methylprednisolone.
Acknowledgements
The authors gratefully acknowledge PCR Laboratories,India for providing necessary facilities to carry out this work.
R E F E R E N C E S
[1]Fang J,Sukumaran S,Dubois DC,et al.Meta-modeling of methylprednisolone effects on glucose regulation in rats. PLoS ONE 2013;8:e81679.
[2]Antal EJ,Wright CE 3rd,Gillespie WR,et al.Infuence on the route of administration on the pharmacokinetics of methylprednisolone.J Pharmacokinet Biopharm 1983;11:561–563.
[3]Hu X,Zheng Y,Wu G,et al.An economical online solidphase extraction LC–MS/MS method for quantifying methylprednisolone.J Chromatogr Sci 2014;53(6):1013–1019.
[4]Difrancesco R,Frerichs V,Donnelly J,et al.Simultaneous determination of cortisol,dexamethasone, methylprednisolone,prednisone,prednisolone, mycophenolic acid and mycophenolic acid glucuronide in human plasma utilizing liquid chromatography–tandem mass spectrometry.J Chromatogr B 2007;859:42–51.
[5]T?lgyesi A,Sharma VK,Fekete S,et al.Simultaneous determination of eight corticosteroids in bovine tissues using liquid chromatography–tandem mass spectrometry. J Chromatogr B 2012;906:75–84.
[6]Chen D,Tao Y,Liu Z,et al.Development of a liquid chromatography–tandem mass spectrometry with pressurized liquid extraction for determination of glucocorticoid residues in edible tissues.J Chromatogr B 2011;879:174–180.
[7]Chen D,Tao Y,Liu Z,et al.Development of a liquid chromatography–tandem mass spectrometry(LC–MS/MS) method for the quantifcation of glucocorticoid residues in edible tissues of swine,cattle,sheep,and chicken.Food Addit Contam Part A Chem Anal Control Expo Risk Assess 2010;27:1363–1371.
[8]Pozo OJ,Marcos J,Matabosch X,et al.Using complementary mass spectrometric approaches for the determination of methylprednisolone metabolites in human urine.Rapid Commun Mass Spectrom 2012;26:541–553.
[9]Vree TB,Maljers L,Van den Borg N,et al.High-performance liquid-chromatographic-atmospheric-pressure chemicalionization ion-trap mass-spectrometric identifcation of isomeric C6-hydroxy and C20-hydroxy metabolites of methylprednisolone in the urine of patients receiving highdose pulse therapy.J Pharm Pharmacol 1999;51:1155–1166.
[10]T?lgyesi A,Sharma VK,Fekete J.Development and validation of a method for determination of corticosteroidsin pig fat using liquid chromatography–tandem mass spectrometry.J Chromatogr B 2011;879:403–410.
[11]Malone EM,Elliott CT,Kennedy DG,et al.Screening and quantitative confrmatory method for the analysis of glucocorticoids in bovine milk using liquid chromatography–tandem mass spectrometry.J AOAC Int 2010;93:1656–1665.
[12]T?lgyesi A,T?lgyesi L,Sharma VK,et al.Quantitative determination of corticosteroids in bovine milk using mixed-mode polymeric strong cation exchange solidphase extraction and liquid chromatography–tandem mass spectrometry.J Pharm Biomed Anal 2010;53: 919–928.
[13]Zhang SQ,Thorsheim HR,Penugonda S,et al.Liquid chromatography-tandem mass spectrometry for the determination of methylprednisolone in rat plasma and liver after intravenous administration of its liver-targeted dextran prodrug.J Chromatogr B Analyt Technol Biomed Life Sci 2009;877:927–932.
[14]US DHHS,FDA,CDER.Guidance for industry:bioanalytical method validation.Rockville,MD:US Department of Health and Human Services,Food and Drug Administration,Center for Drug Evaluation and Research and Center for Veterinary Medicine;2001.
[15]Science and Medicinal Health,European Medicines Agency (EMEA).Guideline on bioanalytical method validation, EMEA/CHMP/EWP/192217/2009;21 July 2011.
[16]Viswanathan CT,Bansal S,Booth B,et al.Quantitative bioanalytical methods validation and implementation:best practices for chromatographic and ligand binding assays. Pharm Res 2007;24:1962–1973.
[17]De Boer T,Wieling J.Incurred sample accuracy assessment: design of experiments based on standard addition. Bioanalysis 2011;3:983–992.
*< class="emphasis_italic">Corresponding author.
.Department of Pharmacy,Shri Jagdishprasad JhabarmalTibrewala University,Jhunjhunu,Rajasthan 333001,India. Tel.:+91 9030005753;fax:+91 4065742040.
E-mail address:lalprasanth@yahoo.com(M.L.Lal Prasanth).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2015.06.005
1818-0876/?2016 Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
 Asian Journal of Pharmacentical Sciences2016年3期
Asian Journal of Pharmacentical Sciences2016年3期
- Asian Journal of Pharmacentical Sciences的其它文章
- Formulation and evaluation of co-prodrug of furbiprofen and methocarbamol
- Auricularia auricular polysaccharide-low molecular weight chitosan polyelectrolyte complex nanoparticles:Preparation and characterization
- Brain targeted delivery of paclitaxel using endogenous ligand
- Generic sustained release tablets of trimetazidine hydrochloride:Preparation and in vitro – in vivo correlation studies
- Effect of process and formulation variables on the preparation of parenteral paclitaxel-loaded biodegradable polymeric nanoparticles: A co-surfactant study
- Preparation and evaluation of PEGylated phospholipid membrane coated layered double hydroxide nanoparticles