
Cortical spreading depression-induced preconditioning in the brain
2016-02-09 05:17:24PingpingShenShuaiHouDiMaMingmingZhaoMingqinZhuJingdianZhangLiangshuFengLiCuiJiachunFengInstituteofNeuroscienceCenterandNeurologyDepartmenttheFirstAffiliatedHospitalofJilinUniversityChangchunJilinProvinceChina
中國神經再生研究(英文版) 2016年11期
Ping-ping Shen, Shuai Hou, Di Ma, Ming-ming Zhao, Ming-qin Zhu, Jing-dian Zhang, Liang-shu Feng, Li Cui, Jia-chun FengInstitute of Neuroscience Center and Neurology Department, the First Affiliated Hospital of Jilin University, Changchun, Jilin Province, China
REVIEW
Cortical spreading depression-induced preconditioning in the brain
Ping-ping Shen, Shuai Hou, Di Ma, Ming-ming Zhao, Ming-qin Zhu, Jing-dian Zhang, Liang-shu Feng, Li Cui*, Jia-chun Feng*
Institute of Neuroscience Center and Neurology Department, the First Affiliated Hospital of Jilin University, Changchun, Jilin Province, China
How to cite this article:Shen PP, Hou S, Ma D, Zhao MM, Zhu MQ, Zhang JD, Feng LS, Cui L, Feng JC (2016) Cortical spreading depression-induced preconditioning in the brain. Neural Regen Res 11(11):1857-1864.
Open access statement:This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
Funding:This research was supported by the National Natural Science Foundation of China, No. H0906-C090201; a grant from the National Science and Technology Support Program of China, No. 3G013F843428.
Cortical spreading depression is a technique used to depolarize neurons. During focal or global ischemia, cortical spreading depression-induced preconditioning can enhance tolerance of further injury. However, the underlying mechanism for this phenomenon remains relatively unclear. To date, numerous issues exist regarding the experimental model used to precondition the brain with cortical spreading depression, such as the administration route, concentration of potassium chloride, induction time, duration of the protection provided by the treatment, the regional distribution of the protective effect, and the types of neurons responsible for the greater tolerance. In this review, we focus on the mechanisms underlying cortical spreading depression-induced tolerance in the brain, considering excitatory neurotransmission and metabolism, nitric oxide, genomic reprogramming, inflammation, neurotropic factors, and cellular stress response. Specifically, we clarify the procedures and detailed information regarding cortical spreading depression-induced preconditioning and build a foundation for more comprehensive investigations in the field of neural regeneration and clinical application in the future.
nerve regeneration; cortical spreading depression; neuronal depolarization; ischemic tolerance; peri-infarct depolarization; excitatory neurotransmission; nitric oxide; genomic reprogramming; inflammation; neurotropic factors; cellular stress response; neural regeneration
Introduction
Ischemic brain injuries that result from blood perfusion and blood-flow reduction are a wide-spread cause of death and disability worldwide (Burda et al., 2014; Zhao et al., 2014; Weaver and Liu, 2015). Conditioning methods comprise the most comprehensively studied strategies aimed at overcoming these injuries. Preconditioning refers to procedures that provide neuroprotection against the harmful effects of subsequent, prolonged, lethal ischemia. This process involves applying a detrimental stimulus close to the threshold of damage to the brain (Ding et al., 2016). Endogenous protection seems to rely on the degree of preconditioning. The sub-toxic level stimuli used in preconditioning range from brief ischemic episodes and chemical stimulation to cortical spreading depression (CSD) (Han et al., 2014; Kristensen and Ropcke, 2015). CSD comprises a non-physiological, global depolarization of neurons and astrocytes that may be initiated in the cortex of a normally perfused brain (Tang et al., 2014). The propagation velocity of CSD is 2–5 mm/min and is followed by remarkable increases in cerebral blood flow, oxygenation of tissue, and metabolic rate. However, these changes are only transient (Passaro et al., 2010; Rana et al., 2012).
Kobayashi et al. (1995) first demonstrated that CSD induced ischemic tolerance in the hippocampal CA1 neurons of rats. To date, several other studies of cortical neurons have replicated these results and further characterized the protective effects of CSD (Kobayashi et al., 1995; Matsushima et al., 1996). Extensive animal studies of injury models suggest that the brain may be preconditioned by CSD to resist acute injuries such as middle cerebral artery occlusion, two-vessel occlusion, three-vessel occlusion, and oxygen-glucose deprivation (Taga et al., 1997; Koroleva et al., 1998; Horiguchi et al., 2006). The concept of utilizing CSD as a preconditioning method originated from investigations of minor cortical injury-induced tolerance against ischemic insults (Sharp et al., 1990; Koroleva et al., 1998). CSD is one way to transiently depolarize neurons (Taga et al., 1997; Muramatsu et al., 2004). Several decades ago, CSD drew public attention because of its relevance for multiple pathophysiological processes such as subarachnoid hemorrhage (Seule et al., 2015), amnesia (Maggioni et al., 2011), traumatic brain injury (Hartings et al., 2014), cerebral hypoxia and stroke (Risher et al., 2012; Bere et al., 2014), and the aura of migraine (Nozari et al., 2010; Shyti et al., 2015).
Evidence indicates that CSD preconditioning is complex and involves multiple effectors, such as metabolic inhibition, a shift in neuronal excitatory/inhibitory balance, changes in inflammatory flow, and stress-related protein alterations (van Rensburg et al., 2009; Passaro et al., 2010). However, the precise mechanism through which CSD preconditioning induces tolerance is still unknown. Nevertheless,CSD preconditioning and ischemic preconditioning share common features, and thus, substantial work regarding the mechanism of CSD preconditioning was initially derived from ischemic preconditioning.
In this review, we focus on the experimental evidence for CSD preconditioning in the brain and systematically survey the literature regarding its mechanism with a goal towards developing paradigms for neuroprotection.
Experimental Evidence of CSD Preconditioning
Experimental methods
The many models of CSD preconditioning are characterized by substantial inconsistencies in induction measures, induction period, and experimental materials, which lack standards. One goal of this review is to clarify the differences in model development. Models for CSD preconditioning can be established bothin vivoandin vitro(Viggiano et al., 2008, 2014). Mice and rats are both suitable for CSD inductionin vivo(Pietrobon and Moskowitz, 2014), while brain slices are commonly used forin vitroinvestigations (Gniel and Martin, 2013). Furthermore, cell cultures, such as those of rat primary cortical neurons (Schock et al., 2007, 2008) and hippocampal organotypic cultures (Kunkler et al., 2004), have both been validated as experimental models for CSD-induced preconditioning. Of these different approaches, rats are the most similar to humans in cerebral vascular anatomy and brain physiology.
Experimental induction of CSD is accomplishedviaapplying potassium chloride (KCl) to the cortical surface (Somjen, 2001). It may also be inducedviamicroinjection of KCl or potassium acetate (Amemori and Bures, 1990) into the frontal cortex (CSD hemisphere). Other methods include needle prick (Taga et al., 1997), mechanical trauma (Mun-Bryce et al., 2006), and electrical stimulation (Martens-Mantai et al., 2014). Furthermore, to induce a prolonged period of CSD, KCl may be appliedviaan osmotic pump (Yanamoto et al., 2000a). However, systematic investigation has not been conducted to determine whether the protective effects provided by CSD preconditioning differ depending on the induction method. Matsushima et al. (1998) and Yanamoto et al. (2004) suggested that CSD may exert neuroprotective effects when KCl is applied epidurally rather than intra-cortically. It could be concluded that intra-cortical administration of KCl may cause injury to the cortex, and could subsequently affect the results.
KCl concentration and time course of preconditioning
The literature regarding the best KCl concentration for CSD preconditioning may cause confusion. Here, we summarize the different KCl concentrations that have been tested and provide guidance for selecting appropriate experimental methods. As shown inTable 1, the concentration used to induce CSD waves ranges from 0.5 M to 5 M, with 2 M KCl being the most common. A high concentration of KCl (5 M) has been reported to cause tiny cortical lesions, and may influence the results (Muramatsu et al., 2004). In contrast, low concentration of KCl, such as 0.5 M, may not be sufficient for generating a significant burden on ionic homeostasis in the extracellular environment, and may therefore fail to induce CSD. So, our group has utilized 1 M KCl to induce CSD preconditioning.
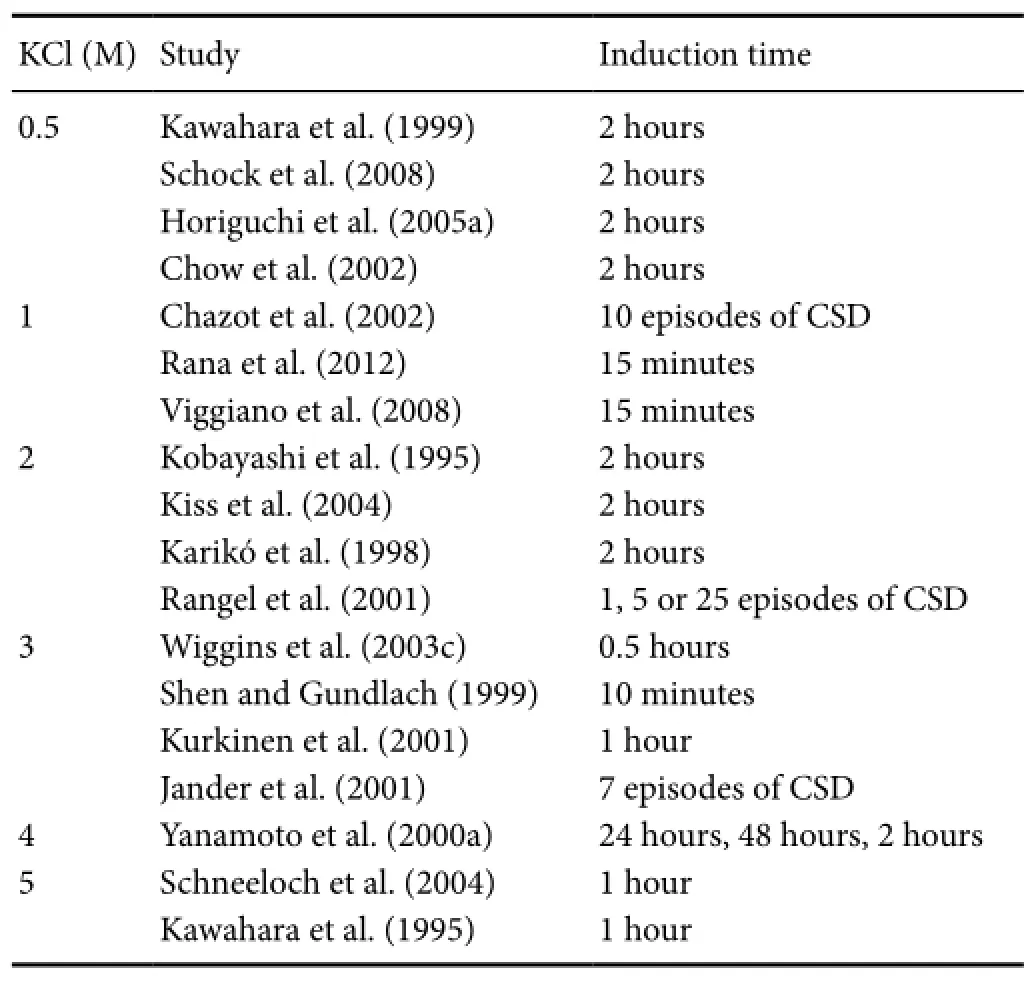
Table 1 Potassium chloride (KCl) concentration and induction time in cortical spreading-depression (CSD) preconditioning
Tolerance can be achieved through two mechanistically different processes, depending on whether the preconditioning induction period is brief or prolonged. Brief preconditioning refers to time course that ranges from several minutes (Wiggins et al., 2003a; Schock et al., 2008) to several hours (Kawahara et al., 1995, 1999), and is typically two hours (Kobayashi et al., 1995; Kiss et al., 2004; Horiguchi et al., 2005a, b). Prolonged preconditioning indicates a time course lasting 1–2 days and is inducedviaan intracerebral microinfusion of KCl for 24–48 hours (Yanamoto et al., 2000a, b) (Table 1). The two types of preconditioning share a common biochemical feature that initiates the signaling cascades. The protection conferred by brief preconditioning results from post-translational protein modifications and does not last long (Horiguchi et al., 2005a). Prolonged preconditioning, however, leads to delayed tolerance that is mediated by protein synthesis and can persist for several days (Yanamoto et al., 2000a; Urbach et al., 2006).
Furthermore, although studies have used the number of CSD episodes as a means of unifying the time course of CSD preconditioning, they do not consider the total time of the procedure. For example, Rangel et al. (2001) administered 1, 5, or 25 episodes of CSD to induce ischemic tolerance in the cortex. The authors suggested that the CSD-induced tolerance was probably dose-dependent. Other researchers have subsequently confirmed this point. Jander et al. (2001) recorded 7 direct current deflections (approximately 100 minutes after their application) with 3 M KCl on the cortex, whereas Chazot et al. (2002) elicited 10 full consecutive recurrent CSDs. The link between the number of CSD waves and the degree of CSD-mediated tolerance still needs to be comprehensively investigated. Determining which type of method is best remains difficult, because each approach has its own merits. However,our group tends to support the former approach, in which CSD waves are induced in a defined time period, such as 2 hours, because the CSD waves are not separate from each other. Thus, a single CSD wave is not equal to each other in the influence it exerts on ischemic tolerance.
The protective time window of CSD preconditioning
As demonstrated in multiple series of preconditioning studies, the protective effect of CSD preconditioning is transient and does not last throughout life (Gong et al., 2014; Lee et al., 2015). Reports are inconsistent regarding the duration of the protective time window provided by CSD preconditioning, ranging from several hours to several days. With brief CSD preconditioning, Taga et al. (1997) showed that the tolerance began after 1 day of CSD treatment and was still significant after 7 days. However, tolerance level was not investigated beyond this time point. Kobayashi et al. (1995) and other scholars (Otori et al., 2003; Yanamoto et al., 2004) supported the view that when CSD is induced 1 day before an arterial occlusion, the protective effect can begin in an earlier time window, such as 12 hours after the start of CSD preconditioning (Matsushima et al., 1998). However, Kawahara et al. (1995) found a tolerance effect 3 days after CSD, but not after 1 or 7 days. Furthermore, when preconditioning is prolonged to 24–48 hours of uninterrupted CSD induction, the duration of neuroprotection is also prolonged. A prolonged period of 48-hour CSD preconditioning can induce a potent ischemic tolerance as long as 12–15 days, and correspondingly reduce the infarct volume in ischemic brain injury (Yanamoto et al., 2004). To our knowledge, no reports have investigated longer periods.
Protective effects of CSD preconditioning on different brain regions
The neuroprotective effects of CSD preconditioning are regional, especially in the cortex (Otori et al., 2003). However, the protection provided by CSD-induced tolerance in the hippocampus is controversial. Taga et al. (1997) suggested that CSD-induced ischemic depolarization could keep the cortical neurons from suffering ischemic injury during an infarction in the cortex, but not in subcortical regions or hippocampus. Furthermore, the protective effects of CSD preconditioning on subcortical areas such as the striatum are not dramatically different than those on the hippocampus. Kobayashi et al. (1995) and other researchers (Plumier et al., 1997; Matsushima et al., 1998) reported similar results using a combined method that assessed neuronal necrosis in the CSD-treated ipsilateral brain compared with the contralateral brain. Kiss et al. (2004) confirmed that topical application of 2 M KCl to the cortical surface (successive, unilateral CSDs), could lead to an ipsilateral increase in kynurenate levels, and was considered a neuroprotective effector predominantly in the frontal, parietal, and occipital cortices, but not in other brain regions.
The protection conferred on different layers of the cortex has been considered to be heterogeneous. The expression of phosphor-5′ adenosine monophosphate-activated protein kinase (p-AMPK) induced by CSD preconditioning is mainly in the superficial cortical layers as determined by immunohistochemical and immunofluorescence approaches (Viggiano et al., 2014). Using a mouse neocortical brainslice model, Gniel and Martin (2013) showed that the neuroprotective effect observed after repeated episodes of CSD preconditioning was lamina specific. Their results showed that the effect of CSD-induced preconditioning lasted 15 to 30 minutes in layer V, while no neuroprotective effect was observed in layer II/III pyramidal neurons.
Kawahara et al. (1999) and Plumier et al. (1997) have confirmed that CSD preconditioning also provides protection in the hippocampus, however, they did not determine whether the extent of this effect is the same as that in the cortex.
Protective effects of CSD preconditioning on different cell types
It is commonly accepted that the adaptive response induced by CSD preconditioning occurs preferentially in cortical neurons (Taga et al., 1997). Kawahara et al. (1997) suggested that CSD can protect neurons from severe ischemic injury in both the hippocampus and the cortex. Another study has indicated that an increase in phosphor-adenosine 5′-monophosphate-activated protein kinase α, a protective factor in CSD preconditioning, was confined to neurons and was not identified in astroglial cells (Viggiano et al., 2014).
The effect that CSD preconditioning has on astrocytes remains controversial. The most important role of astrocytes is metabolizing and removing glutamate. The major rate-limiting enzyme in metabolizing glutamate is glutamine synthetase, which is primarily derived from astrocytes. Additionally, it has been proved that two functions of reactive astrocytes are to scavenge for glutamate and to protect cortical neurons from ischemic infarction (Zou et al., 2010; Cooper, 2012). The expression of growth factor is strongly associated with CSD-induced ischemic tolerance (Matsushima et al., 1998). Furthermore, Kawahara et al. (1997) showed that heat-shock protein 27 (HSP27) could be activated by CSD preconditioning. Using immunohistochemical analyses, Matsushima et al. (1998) showed that levels of the astrocyte marker glial fibrillary acidic protein (GFAP) increased after CSD preconditioning and peaked at 3 days. GFAP levels in astrocytes have also been shown to increase relative to normal levels following CSD (Yanamoto et al., 2000a). Kawahara et al. (1999) suggested that the resistance to ischemic injury induced by CSD resulted from a glial cell activation, and may indicate a new target for long-term protection of neurons.
CSD was shown to induce the expression of neuronal nitric oxide synthase (nNOS), and the double labeling of nNOS and GFAP identified these nNOS-positive cells as astrocytes (Caggiano and Kraig, 1998). However, both neurons and astrocytes are important for ischemic tolerance and may work togetherviamultiple mechanisms, as evidenced by Matsushima et al. (1998) and other scholars (Wiggins et al., 2003a; Viggiano et al., 2008). These molecular studies have demonstrated that both neuronal and non-neuronal cells (such as reactive astrocytes) increased the expressions of presumed neuroprotective proteins following CSD preconditioning (Wiggins et al., 2003a).
Mechanism of CSD Preconditioning
Excitatory neurotransmission and metabolism
Changes excitatory neurotransmission and metabolic down-regulation are primary mechanisms of CSD-inducted ischemic tolerance in invertebrate species. Neuronal excitotoxicity (Fiszman et al., 2010), such as that caused by glutamate, is a significant source of the pathophysiology seen during ischemic injury. Ischemic tolerance induced by CSD may occur through reduced glutamate release or through greater uptake (Schock et al., 2007). This finding indicates that glutamate mechanism is vital to ischemic tolerance induced by CSD. Excitotoxicity and excitatory pathways have been shown to become suppressed through down-regulation of aminomethyl phosphonic acid (AMPA) and the N-methyl-D-aspartate (NMDA) receptors during CSD preconditioning (Lindquist and Shuttleworth, 2014). The major AMPA and NMDA receptor subunits in the mammalian cortex include GluR1 and GluR2 and NR1, NR2A, and NR2B (Lussier et al., 2015). Chazot et al. (2002) demonstrated that delayed changes in AMPA and α7 nicotinic acetylcholine receptor protein expression may contribute to the increased tolerance associated with CSD.
Extracellular glutamate is cleared primarily through excitatory amino acid transporters (EAATs) that uptake glutamate in the cortex.(Mulligan and Mindell, 2013). EAAT1 and EAAT2 are astrocyte-derived (Gras et al., 2012; Fahlke et al., 2016), whereas EAAT3 is primarily derived from neuron. Douen et al. (2000) indicated that CSD preconditioning could affect levels of EAAT1 and EAAT2, but not NMDA receptors.
In the brain, adenosine triphosphate (ATP) acts as a signaling molecule, and modifies cellular differentiation and proliferation (Rodrigues et al., 2015). ATP also modulates neuronal excitability and alters gene expression (Wang et al., 2015). During CSD, the most prominent phenomenon is the transient but substantial decrease in ATP levels caused by increased metabolic activity essential for membrane potential restoration (Burnstock and Knight, 2004). This leads to a dramatic release of ATP into the extracellular space, and subsequent activation of purinergic receptors, which are important for ischemic tolerancein vivo(Schock et al., 2007) andin vitro(Schock et al., 2008). Moreover, depolarization causes ATP release into the extracellular medium. Several purinergic receptors are purported to be involved in the induction of ischemic tolerance. Among these, the P2Y receptor appears to be the most important. At the same time, adenosine or glutamate receptor activation is independent of ischemic tolerance (Burnstock, 2004). CSD preconditioning is thought to modify the expression of genes that reduce ATP use, thereby reducing energy metabolism (Horiguchi et al., 2005b).
Nitric oxide
Nitric oxide signaling has been comprehensively studied with regards to its transmission between neurocytes and to cell survival/toxic conditions in ischemic brain injury (Zheng et al., 2016). Under certain conditions, nitric oxide is very important in mediating neuroprotective effects. Numerous studies have proven the crucial role of the nitric oxide-generating pathway in establishing resistance to cerebral ischemiaviaother preconditioning methods, such as oxygen-glucose deprivation, and hypoxia (Arabian et al., 2015). However, the role of nitric oxide in CSD preconditioning remains unclear. CSD preconditioning could generate high levels of cortical nitric oxide in a rapidly activated and chronically up-regulated manner (Read et al., 1997). There are three types of nitric oxide synthase (NOS) that act to synthesize nitric oxide (Kolár and Nohejlová, 2014): neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS). Caggiano and Kraig (1998) revealed a vital connection between nNOS levels and the CSD-induced resistance against ischemic injury in the brain. Furthermore, the identity of cells that express NOS after cortical depression has spread is important, because the changes in intracellular environments are heterogeneous and cell type-specific, and may control NOS activity (Caggiano and Kraig, 1998). Viggiano et al. (2008) performed time-course experiments in which CSD selectively increased the expression of some nNOS activity-related genes. Wiggins et al. (2003a) suggested that both transcriptional and translational changes occur after CSD preconditioning with regards to nNOS adaptor proteins, and these changes were able to modify nitric oxide levels and nNOS activity. The nNOS adaptor proteins, including the carboxy-terminal PDZ ligand of nNOS, protein inhibitor of nNOS, postsynaptic density-95, manganese superoxide dismutase, and copper/ zinc-superoxide dismutase, have been proven to be related to the ischemic tolerance following CSD.
Genomic reprogramming
CSD-induced ischemic tolerance is accompanied by many changes in gene expression. Thus, the fundamental genomic reprogramming of cells stimulated by CSD preconditioning may confer the cytoprotective effect (Passaro et al., 2010; Rana et al., 2012). Although it is unlikely that all of these genes are of equal importance in the induction of ischemic tolerance, many factors are involved in changing the brain to a more tolerant phenotype (Tang et al., 2006).
CSD preconditioning-induced ischemic tolerance can regulate genes involved in metabolic pathways (Douen et al., 2000), inflammatory responses (Jander et al., 2001), nitric oxide synthesis and metabolism (Passaro et al., 2010) including NOS (Shen and Gundlach, 1999), stress response (Dietrich et al., 2000; Rangel et al., 2001), neurotropic factors (Karikó et al., 1998; Badisco et al., 2011), calcium-independent protein kinase C (Koponen et al., 1999), mitogen-activated protein kinase phosphatase-1 (Herdegen et al., 1993), junB, c-jun, and c-fos (Hermann et al., 1999). The CSD preconditioning-induced genomic response represents the complex interplay between different signaling pathways. However, the molecular mechanisms remain unclear. CSD preconditioning could induce specific alterations in gene expression rather than random changes.
Lysine methylation is also involved in the effects of CSD preconditioning (Passaro et al., 2010). CSD preconditioning was shown to regulate gene expression through epigenetic modifications (Passaro et al., 2010), such as the induction of histone H3 lysine 4 dimethylation and lysine 9 dimethylation of the H3 histone. The major characteristics of epigenetic modification are that it does not alter the total DNAsequence and it involves interdependent mechanisms, such as histone modifications. The most prominent signature of CSD-induced epigenetic modifications is heritable (van der Maarel, 2008). Abnormal epigenetic modification could be proposed as an indicator of various pathological states. Equally likely however, is that epigenetic gene regulation could be used therapeutically on specific targets (Kelly et al., 2010). Epigenetic modifications of gene expression may be influenced by CSD and may be a pivotal molecular mechanism through which ischemic tolerance is induced by CSD preconditioning (Kelly et al., 2010; Passaro et al., 2010).
Inflammatory mechanisms
Inflammatory responses are the most extensively studied effects among all preconditioning models, including CSD preconditioning. CSD imparts a certain degree of ischemic tolerance to the brain and may have multiple effects on the expression of inflammatory genes (Thompson and Hakim, 2005). CSD modifies components of the inflammatory cascade, with some inflammatory mediators being detrimental and others being beneficial to the progression of ischemic injury (Thompson and Hakim, 2005).
Cytokines can regulate the prognosis of severe ischemic brain damage. Cytokines are pleiotropic in combination with other cytokines (Xu et al., 2011), however, their roles in neuroprotection remain incompletely defined. Some authors have characterized the magnitude, time course, and diversity of cytokine responses to CSD (Jander et al., 2001). A pharmacological study demonstrated that depending on the time window and dosage, cytokines may also confer protection against neuronal injuryin vitroafter CSD induction in the rat brain (Jander et al., 2001).
In CSD preconditioning and other preconditioning methods, the expression of cytokines partly represents the physiological stress response, which is essential in ischemic tolerance (Jander et al., 2001). Furthermore, levels of cyclooxygenase-2 were shown to be related to CSD-induced ischemic tolerance (Horiguchi et al., 2006).
In general, a comprehensive understanding of inflammatory processes influenced by CSD contributes to the recognition of novel targets for acute treatment of stroke (Thompson and Hakim, 2005).
Neurotropic factors
Neurotrophic factors refer to polypeptides that promote cell survival, growth, and differentiation through regulation of molecular signals. CSD-induced ischemic tolerance could provide long-lasting neuroprotection by activating glial cells and up-regulating trophic factors (Kawahara et al., 1999).
The most commonly investigated trophic factor is brain-derived neurotropic factor (BDNF). Substantial evidence has confirmed that CSD preconditioning increases BDNF protein (Kawahara et al., 1999; Rangel et al., 2001). BDNF was shown as part of the mechanism underlying ischemic tolerance following preconditioning with repetitive CSD (Matsushima et al., 1998; Yanamoto et al., 2000b). Furthermore, changes in BDNF expression as well as mRNA in distinct regions of the cerebral cortex exhibited different characteristics.
Sight increases in BDNF levels were observed in the cortex after 1 day and from days 3 to 7 days after CSD preconditioning, while a delayed decrease in levels was observed in the hippocampus independent of BDNF mRNA levels (Kawahara et al., 1997). At the same time, BDNF-like protein in cell nuclei were able to induce ischemic tolerance in the brain (Yanamoto et al., 2000a, 2004). Other neurotropic factors include basic fibroblast growth factor, and GFAP. It has been shown that up-regulation of the glial cell-marker GFAP can induce long-term protection in the central nervous system (Matsushima et al., 1998; Kawahara et al., 1999).
Cellular stress response
The unfolded protein response and the heat-shock response are two primary stress targets which are defense reactions to conditions associated with cytoplasmic or endoplasmic reticulum dysfunction. These responses represent defensive reactions to endoplasmic reticulum dysfunction. The ischemic tolerance conferred by CSD preconditioning has been associated with the unfolded protein response (Schneeloch et al., 2004).
HSPs are a set of highly conserved proteins which are upregulated by ischemic and oxidative injury or other kinds of stresses. HSPs comprise a wide range of stress proteins that increase the ability of cells to resist different kinds of stress, including that produced by ischemia (Parsell and Lindquist, 1993). Thus, the expression of HSPs and related stress proteins is an important mechanism by which tolerance against ischemia is induced, especially in CSD-induced preconditioning. There are many types of HSPs, including HSP73, HSP72, HSP70 and HSP27, which are extensively studied in CSD preconditioning. Karikó et al. (1998) indicated that after CSD induction, levels of HSP73 mRNA were elevated at 2 and 24 hours. Regarding HSP72, most studies indicate that CSD did not increase hsp72 mRNA or HSP72 protein expression in cortex (Amemori and Bures, 1990; Karikó et al., 1998; Matsushima et al., 1998). However, Kobayashi et al. (1995) found small increases in hsp72 mRNA levels following 25 episodes of CSD, suggesting that a distinct threshold in the number of CSDs exists for hsp72 induction. In contrast, Yanamoto et al. (2000a) demonstrated that HSP70 did not participate in the neuroprotective effect of CSD preconditioning. Hsp27 might be induced by cerebral focal/global ischemia (Kato et al., 1995). Hsp27 might be expressed in astrocytes after cortical application of potassium chloride (Plumier et al., 1997). Moreover, hsp27 mRNA levels were elevated in astrocytes, and peaked at 1 and 6 days after CSD. However, no hsp70 expression was found during CSD episodes (Yanamoto et al., 2000a).
Other factors
There are several other aspects concerning CSD-induced tolerance. The expression of fos or tissue plasminogen activator may be associated with ischemic tolerance (Karikó et al., 1998). Clusterin, also known as sulfated glycoprotein-2 and apolipoprotein J, is a major secretory glycoprotein participating in neuronal tolerance (Wilson and Easterbrook-Smith, 2000; Wiggins et al., 2003b). Furthermore, several protein kinases involved in all aspects of molecular biology, cellular metabolism, and signal transduction, have been associated withCSD preconditioning, including protein kinase C (Kurkinen et al., 2001), adenosine 5′-monophosphate-activated protein kinase (Viggiano et al., 2014), and mitogen-activated protein kinase (Chow et al., 2002).
Clinical Translation of CSD Preconditioning
As an extensively researched animal model for inducing ischemic tolerance (Shpargel et al., 2008), CSD preconditioning is also faced with problems transforming basic research into clinical treatments, an issue for other preconditioning methods as well, such as ischemic preconditioning and hyperbaric oxygen preconditioning. Although numerous studies have clearly indicated the neuroprotective effects of the preconditioning method, translation into clinical usage is still disappointing. In actuality, we cannot use the same direct means in human cortex as is used in experimental methods with animals. Studies regarding the preconditioning effects of CSD could broaden our insights into its neuroprotective aspects because of the resemblance between CSD waves and peri-infarct depolarizations (Gorji, 2001; Ayata and Lauritzen, 2015). CSD is more suitable for investigating the pathological and physiological evolution of ischemic penumbra and therefore seeking new therapeutic targets for neurovascular disease treatment. Lately, the potential of CSD for clinical translation has become more and more feasible (Kramer et al., 2016). Meanwhile, the electrophysiological characteristics of CSD waves have drawn much attention of clinical researchers. Depolarization/depression-induced electrocorticogram fluctuations are being recorded more and more often and used as biomarkers during neurocritical care monitoring neurosurgical operations, and may provide a diagnostic measure that summarizes metabolic failure and excitotoxic injury (Hartings et al., 2016).
Conclusions
CSD preconditioning may represent a powerful approach to investigate neuroprotection for cerebral ischemia. Moderate concentrations of KCl, such as 1–2 M, are most suitable for inducing CSD preconditioning, and a time-quantified, rather than number-quantified, induction period is more reasonable. Furthermore, the protective effect of CSD preconditioning does not last long, ranging from several minutes to a maximum of one month. Moreover, CSD-induced tolerance predominately occurs in the cortex (where it is lamina-specific), with some tolerance in the hippocampus and subcortical regions. Neurons and astrocytes are both important for inducing the preconditioning effect.
As a result of its similarity with peri-infarct depolarization, which exhibits prolonged direct current shifts and causes progressive recruitment of the penumbra into the infarct core, a comprehensive understanding of the methodology and mechanism of CSD preconditioning will facilitate therapeutic strategies for acute ischemic events. Improved overall understanding of CSD preconditioning should facilitate the identification of inherent correlations between CSD and peri-infarct depolarization, and may prevent or at least reduce neuronal damage.
Author contributions:PPS, LC and JCF conceived and designed the work. SH, DM, MMZ, JDZ and LSF prepared and edited the paper. MQZ reviewed the paper. All authors approved the final version of the paper.
Conflicts of interest:None declared.
Plagiarism check:This paper was screened twice using CrossCheck to verify originality before publication.
Peer review:This paper was double-blinded and stringently reviewed by international expert reviewers.
Amemori T, Bures J (1990) Ketamine blockade of spreading depression: rapid development of tolerance. Brain Res 519:351-354.
Arabian M, Aboutaleb N, Soleimani M, Mehrjerdi FZ, Ajami M, Pazoki-Toroudi H (2015) Role of morphine preconditioning and nitric oxide following brain ischemia reperfusion injury in mice. Iran J Basic Med Sci 18:14-21.
Ayata C, Lauritzen M (2015) Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 95:953-993.
Badisco L, Ott SR, Rogers SM, Matheson T, Knapen D, Vergauwen L, Verlinden H, Marchal E, Sheehy MR, Burrows M, Broeck JV (2011) Microarray-based transcriptomic analysis of differences between longterm gregarious and solitarious desert locusts. PLoS One 6:e28110.
Bere Z, Obrenovitch TP, Kozák G, Bari F, Farkas E (2014) Imaging reveals the focal area of spreading depolarizations and a variety of hemodynamic responses in a rat microembolic stroke model. J Cereb Blood Flow Metab 34:1695-1705.
Burda R, Danielisova V, Gottlieb M, Nemethova M, Bonova P, Matiasova M, Morochovic R, Burda J (2014) Delayed remote ischemic postconditioning protects against transient cerebral ischemia/reperfusion as well as kainate-induced injury in rats. Acta Histochem 116:1062-1067.
Burnstock G (2004) Introduction: P2 receptors. Curr Top Med Chem 4:793-803.
Burnstock G, Knight GE (2004) Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol 240:31-304.
Caggiano AO, Kraig RP (1998) Neuronal nitric oxide synthase expression is induced in neocortical astrocytes after spreading depression. J Cereb Blood Flow Metab 18:75-87.
Chazot PL, Godukhin OV, McDonald A, Obrenovitch TP (2002) Spreading depression-induced preconditioning in the mouse cortex: differential changes in the protein expression of ionotropic nicotinic acetylcholine and glutamate receptors. J Neurochem 83:1235-1238.
Chow AK, Thompson CS, Hogan MJ, Banner D, Sabourin LA, Hakim AM (2002) Cortical spreading depression transiently activates MAP kinases. Mol Brain Res 99:75-81.
Cooper AJL (2012) The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem Res 37:2439-2455.
Dietrich WD, Truettner J, Prado R, Stagliano NE, Zhao W, Busto R, Ginsberg MD, Watson BD (2000) Thromboembolic events lead to cortical spreading depression and expression of c-fos, brain-derived neurotrophic factor, glial fibrillary acidic protein, and heat shock protein 70 mRNA in rats. J Cereb Blood Flow Metab 20:103-111.
Ding XD, Zheng NN, Cao YY, Zhao GY, Zhao P (2016) Dexmedetomidine preconditioning attenuates global cerebral ischemic injury following asphyxial cardiac arrest. Int J Neurosci 126:249-256.
Douen AG, Akiyama K, Hogan MJ, Wang F, Dong L, Chow AK, Hakim A (2000) Preconditioning with cortical spreading depression decreases intraischemic cerebral glutamate levels and down-regulates excitatory amino acid transporters EAAT1 and EAAT2 from rat cerebal cortex plasma membranes. J Neurochem 75:812-818.
Fahlke C, Kortzak D, Machtens JP (2016) Molecular physiology of EAAT anion channels. Pflugers Arch 468:491-502.
Fiszman ML, Ricart KC, Latini A, Rodríguez G, Sica RE (2010) In vitro neurotoxic properties and excitatory aminoacids concentration in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Relationship with the degree of certainty of disease diagnoses. Acta Neurol Scand 121:120-126.
Fouillet A, Levet C, Virgone A, Robin M, Dourlen P, Rieusset J, Belaidi E, Ovize M, Touret M, Nataf S, Mollereau B (2012) ER stress inhibits neuronal death by promoting autophagy. Autophagy 8:915-926.
Gniel HM, Martin RL (2013) Cortical spreading depression-induced preconditioning in mouse neocortex is lamina specific. J Neurophysiol 109:2923-2936.
Gong J, Gong S, Zhang M, Zhang L, Hu Y, Liu Y, Li W (2014) Cerebral ischemic preconditioning reduces glutamate excitotoxicity by up-regulating the uptake activity of GLT-1 in rats. Amino Acids 46:1537-1545.
Gorji A (2001) Spreading depression: a review of the clinical relevance. Brain Res Rev 38:33-60.
Gras G, Samah B, Hubert A, Léone C, Porcheray F, Rimaniol AC (2012) EAAT expression by macrophages and microglia: still more questions than answers. Amino Acids 42:221-229.
Han D, Zhang S, Fan B, Wen LL, Sun M, Zhang H, Feng J (2014) Ischemic postconditioning protects the neurovascular unit after focal cerebral ischemia/reperfusion injury. J Mol Neurosci 53:50-58.
Hartings JA, Wilson JA, Hinzman JM, Pollandt S, Dreier JP, DiNapoli V, Ficker DM, Shutter LA, Andaluz N (2014) Spreading depression in continuous electroencephalography of brain trauma. Ann Neurol 76:681-694.
Hartings JA, Shuttleworth CW, Kirov SA, Ayata C, Hinzman JM, Foreman B, Andrew RD, Boutelle MG, Brennan KC, Carlson AP, Dahlem MA, Drenckhahn C, Dohmen C, Fabricius M, Farkas E, Feuerstein D, Graf R, Helbok R, Lauritzen M, Major S, et al. (2016) The continuum of spreading depolarizations in acute cortical lesion development: Examining Le?o’s legacy. J Cereb Blood Flow Metab doi: 10.1177/0271678X16654495.
Herdegen T, Sandkühler J, Gass P, Kiessling M, Bravo R, Zimmermann M (1993) JUN, FOS, KROX, and CREB transcription factor proteins in the rat cortex: Basal expression and induction by spreading depression and epileptic seizures. J Comp Neurol 333:271-288.
Hermann DM, Mies G, Hossmann KA (1999) Expression of c-fos, junB, c-jun, MKP-1 and hsp72 following traumatic neocortical lesions in rats--relation to spreading depression. Neuroscience 88:599-608.
Hoffmann MC, Nitsch C, Scotti AL, Reinhard E, Monard D (1992) The prolonged presence of glia-derived nexin, an endogenous protease inhibitor, in the hippocampus after ischemia-induced delayed neuronal death. Neuroscience 49:397-408.
Horiguchi T, Kis B, Rajapakse N, Shimizu K, Busija DW (2005a) Cortical spreading depression (CSD)-induced tolerance to transient focal cerebral ischemia in halothane anesthetized rats is affected by anesthetic level but not ATP-sensitive potassium channels. Brain Res 1062:127-133.
Horiguchi T, Snipes JA, Kis B, Shimizu K, Busija DW (2005b) The role of nitric oxide in the development of cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats. Brain Res 1039:84-89.
Horiguchi T, Snipes JA, Kis B, Shimizu K, Busija DW (2006) Cyclooxygenase-2 mediates the development of cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats. Neuroscience 140:723-730.
Jander S, Schroeter M, Peters O, Witte OW, Stoll G (2001) Cortical spreading depression induces proinflammatory cytokine gene expression in the rat brain. J Cereb Blood Flow Metab 21:218-225.
Karikó K, Harris VA, Rangel Y, Monica E, Welsh FA (1998) Effect of cortical spreading depression on the levels of mrna coding for putative neuroprotective proteins in rat brain. J Cereb Blood Flow Metab 18:1308-1315.
Kato H, Kogure K, Liu XH, Araki T, Kato K, Itoyama Y (1995) Immunohistochemical localization of the low molecular weight stress protein HSP27 following focal cerebral ischemia in the rat. Brain Res 679:1-7.
Kawahara N, Ruetzler CA, Klatzo I (1995) Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol Res 17:9-16.
Kawahara N, Croll SD, Wiegand SJ, Klatzo I (1997) Cortical spreading depression induces long-term alterations of BDNF levels in cortex and hippocampus distinct from lesion effects: implications for ischemic tolerance. Neurosci Res 29:37-47.
Kawahara N, Ruetzler CA, Mies G, Klatzo I (1999) Cortical spreading depression increases protein synthesis and upregulates basic fibroblast growth factor. Exp Neurol 158:27-36.
Kelly TK, De Carvalho DD, Jones PA (2010) Epigenetic modifications as therapeutic targets. Nat Biotechnol 28:1069-1078.
Kiss C, Shepard PD, Bari F, Schwarcz R (2004) Cortical spreading depression augments kynurenate levels and reduces malonate toxicity in the rat cortex. Brain Res 1002:129-135.
Kobayashi S, Harris VA, Welsh FA (1995) Spreading depression induces tolerance of cortical neurons to ischemia in rat brain. J Cereb Blood Flow Metab 15:721-727.
Kolár M, Nohejlová K (2014) The role of nitric oxide and NO-synthase in the pathogenesis of cerebral damage after subarachnoid hemorrhage; laboratory models of subarachnoid hemorrhage. Cesk Fysiol 63:34-41.
Koponen S, Kein?nen R, Roivainen R, Hirvonen T, N?rhi M, Chan PH, Koistinaho J (1999) Spreading depression induces expression of calcium-independent protein kinase C subspecies in ischaemia-sensitive cortical layers: regulation by N-methyl-d-aspartate receptors and glucocorticoids. Neuroscience 93:985-993.
Koroleva VI, Korolev OS, Loseva E, Bures J (1998) The effect of MK-801 and of brain-derived polypeptides on the development of ischemic lesion induced by photothrombotic occlusion of the distal middle cerebral artery in rats. Brain Res 786:104-114.
Kramer DR, Fujii T, Ohiorhenuan I, Liu CY (2016) Cortical spreading depolarization: Pathophysiology, implications, and future directions. J Clin Neurosci 24:22-27.
Kristensen SD, Ropcke DM (2015) Ischemic preconditioning and thrombosis. Vasa 44:243-244.
Kunkler PE, Hulse RE, Kraig RP (2004) Multiplexed cytokine protein expression profiles from spreading depression in hippocampal organotypic cultures. J Cereb Blood Flow Metab 24:829-839.
Kurkinen K, Kein?nen R, Li W, Koistinaho J (2001) Preconditioning with spreading depression activates specifically protein kinase Cdelta. Neuroreport 12:269-273.
Lee JC, Kim IH, Park JH, Ahn JH, Cho JH, Cho GS, Tae HJ, Chen BH, Yan BC, Yoo KY, Choi JH, Lee CH, Hwang IK, Cho JH, Kwon YG, Kim YM, Won MH (2015) Ischemic preconditioning protects hippocampal pyramidal neurons from transient ischemic injury via the attenuation of oxidative damage through upregulating heme oxygenase-1. Free Radic Biol Med 79:78-90.
Lindquist BE, Shuttleworth CW (2014) Spreading depolarization-induced adenosine accumulation reflects metabolic status in vitro and in vivo. J Cereb Blood Flow Metab 34:1779-1790.
Lussier MP, Sanz-Clemente A, Roche KW (2015) Dynamic regulation of n-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by posttranslational modifications. J Biol Chem 290:28596-28603.
Maggioni F, Mainardi F, Bellamio M, Zanchin G (2011) Transient global amnesia triggered by migraine in monozygotic twins. Headache 51:1305-1308.
Martens-Mantai T, Speckmann EJ, Gorji A (2014) Propagation of cortical spreading depression into the hippocampus: The role of the entorhinal cortex. Synapse doi: 10.1002/syn.21769.
Matsushima K, Hogan MJ, Hakim AM (1996) Cortical spreading depression protects against subsequent focal cerebral ischemia in rats. J Cereb Blood Flow Metab 16:221-226.
Matsushima K, Schmidt-Kastner R, Hogan MJ, Hakim AM (1998) Cortical spreading depression activates trophic factor expression in neurons and astrocytes and protects against subsequent focal brain ischemia. Brain Res 807:47-60.
Mulligan C, Mindell JA (2013) Mechanism of transport modulation by an extracellular loop in an archaeal excitatory amino acid transporter (EAAT) homolog. J Biol Chem 288:35266-35276.
Mun-Bryce S, Roberts L, Bartolo A, Okada Y (2006) Transhemispheric depolarizations persist in the intracerebral hemorrhage swine brain following corpus callosal transection. Brain Res 1073-1074:481-490.
Muramatsu H, Karikó K, Welsh FA (2004) Induction of tolerance to focal ischemia in rat brain: dissociation between cortical lesioning and spreading depression. J Cereb Blood Flow Metab 24:1167-1171.
Nozari A, Dilekoz E, Sukhotinsky I, Stein T, Eikermann-Haerter K, Liu C, Wang Y, Frosch MP, Waeber C, Ayata C, Moskowitz MA (2010) Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol 67:221-229.
Otori T, Greenberg JH, Welsh FA (2003) Cortical spreading depression causes a long-lasting decrease in cerebral blood flow and induces tolerance to permanent focal ischemia in rat brain. J Cereb Blood Flow Metab 23:43-50.
Parsell DA, Lindquist S (1993) The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Genet 27:437-496.
Passaro D, Rana G, Piscopo M, Viggiano E, De Luca B, Fucci L (2010) Epigenetic chromatin modifications in the cortical spreading depression. Brain Res 1329:1-9.
Pietrobon D, Moskowitz MA (2014) Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci 15:379-393.
Plumier JC, David JC, Robertson HA, Currie RW (1997) Cortical application of potassium chloride induces the low-molecular weight heat shock protein (Hsp27) in astrocytes. J Cereb Blood Flow Metab 17:781-790.
Rana G, Donizetti A, Virelli G, Piscopo M, Viggiano E, De Luca B, Fucci L (2012) Cortical spreading depression differentially affects lysine methylation of H3 histone at neuroprotective genes and retrotransposon sequences. Brain Res 1467:113-119.
Rangel YM, Karikó K, Harris VA, Duvall ME, Welsh FA (2001) Dose-dependent induction of mRNAs encoding brain-derived neurotrophic factor and heat-shock protein-72 after cortical spreading depression in the rat. Mol Brain Res 88:103-112.
Read SJ, Smith MI, Hunter AJ, Parsons AA (1997) The dynamics of nitric oxide release measured directly and in real time following repeated waves of cortical spreading depression in the anaesthetised cat. Neurosci Lett 232:127-130.
Risher WC, Croom D, Kirov SA (2012) Persistent astroglial swelling accompanies rapid reversible dendritic injury during stroke-induced spreading depolarizations. Glia 60:1709-1720.
Rodrigues RJ, Tomé AR, Cunha RA (2015) ATP as a multi-target danger signal in the brain. Front Neurosci 9:148.
Schneeloch E, Wenkel S, Mies G, Paschen W (2004) Spreading depression activates unfolded protein response. Neurosci Lett 368:37-40.
Schock SC, LeBlanc D, Hakim AM, Thompson CS (2008) ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro. Biochem Biophys Res Commun 368:138-144.
Schock SC, Munyao N, Yakubchyk Y, Sabourin LA, Hakim AM, Ventureyra EC, Thompson CS (2007) Cortical spreading depression releases ATP into the extracellular space and purinergic receptor activation contributes to the induction of ischemic tolerance. Brain Res 1168:129-138.
Seule M, Keller E, Unterberg A, Sakowitz O (2015) The hemodynamic response of spreading depolarization observed by near infrared spectroscopy after aneurysmal subarachnoid hemorrhage. Neurocrit Care 23:108-112.
Sharp JW, Sagar SM, Hisanaga K, Jasper P, Sharp FR (1990) The NMDA receptor mediates cortical induction of fos and fos-related antigens following cortical injury. Exp Neurol 109:323-332.
Shen PJ, Gundlach AL (1999) Prolonged induction of neuronal NOS expression and activity following cortical spreading depression (SD): implications for SD- and NO-mediated neuroprotection. Exp Neurol 160:317-332.
Shpargel KB, Jalabi W, Jin Y, Dadabayev A, Penn MS, Trapp BD (2008) Preconditioning paradigms and pathways in the brain. Cleve Clin J Med 75 Suppl 2:S77-82.
Shyti R, Kohler I, Schoenmaker B, Derks RJ, Ferrari MD, Tolner EA, Mayboroda OA, van den Maagdenberg AM (2015) Plasma metabolic profiling after cortical spreading depression in a transgenic mouse model of hemiplegic migraine by capillary electrophoresis - mass spectrometry. Mol Biosyst 11:1462-1471.
Somjen GG (2001) Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81:1065-1096.
Taga K, Patel Piyush M, Drummond John C, Cole Daniel J, Kelly Paul J (1997) Transient neuronal depolarization induces tolerance to subsequent forebrain ischemia in rats. Anesthesiology 87:918-925.
Tang Y, Pacary E, Fréret T, Divoux D, Petit E, Schumann-Bard P, Bernaudin M (2006) Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: identification of potential neuroprotective candidates for stroke. Neurobiol Dis 21:18-28.
Tang YT, Mendez JM, Theriot JJ, Sawant PM, López-Valdés HE, Ju YS, Brennan KC (2014) Minimum conditions for the induction of cortical spreading depression in brain slices. J Neurophysiol 112:2572-2579.
Thompson CS, Hakim AM (2005) Cortical spreading depression modifies components of the inflammatory cascade. Mol Neurobiol 32:51-57.
Urbach A, Bruehl C, Witte OW (2006) Microarray-based long-term detection of genes differentially expressed after cortical spreading depression. Eur J Neurosci 24:841-856.
van der Maarel SM (2008) Epigenetic mechanisms in health and disease. Ann Rheum Dis 67:iii97-iii100.
van Rensburg R, Errington DR, Ennaceur A, Lees G, Obrenovitch TP, Chazot PL (2009) A new model for the study of high-K+-induced preconditioning in cultured neurones: Role of N-methyl-D-aspartate and α7-nicotinic acetylcholine receptors. J Neurosci Methods 177:311-316.
Viggiano E, Viggiano D, Viggiano A, De Luca B, Monda M (2014) Cortical spreading depression increases the phosphorylation of AMP-activated protein kinase in the cerebral cortex. Neurochem Res 39:2431-2439.
Viggiano E, Ferrara D, Izzo G, Viggiano A, Minucci S, Monda M, De Luca B (2008) Cortical spreading depression induces the expression of iNOS, HIF-1α, and LDH-A. Neuroscience 153:182-188.
Wang ZL, Xu DS, Wang YX, Qin H, Geng D (2015) Effect of single nucleotide polymorphisms in the ATP-binding cassette B1 gene on the clinical outcome of traumatic brain injury. Genet Mol Res 14:10948-10953.
Weaver J, Liu KJ (2015) Does normobaric hyperoxia increase oxidative stress in acute ischemic stroke? A critical review of the literature. Med Gas Res 5:11.
Wiggins AK, Shen PJ, Gundlach AL (2003a) Delayed, but prolonged increases in astrocytic clusterin (ApoJ) mRNA expression following acute cortical spreading depression in the rat: evidence for a role of clusterin in ischemic tolerance. Mol Brain Res 114:20-30.
Wiggins AK, Shen P-J, Gundlach AL (2003b) Delayed, but prolonged increases in astrocytic clusterin (ApoJ) mRNA expression following acute cortical spreading depression in the rat: evidence for a role of clusterin in ischemic tolerance. Mol Brain Res 114:20-30.
Wiggins AK, Shen PJ, Gundlach AL (2003c) Neuronal-NOS adaptor protein expression after spreading depression: implications for NO production and ischemic tolerance. J Neurochem 87:1368-1380.
Wilson MR, Easterbrook-Smith SB (2000) Clusterin is a secreted mammalian chaperone. Trends Biochem Sci 25:95-98.
Xu B, Gao X, Xu J, Lei S, Xia ZY, Xu Y, Xia Z (2011) Ischemic postconditioning attenuates lung reperfusion injury and reduces systemic proinflammatory cytokine release via heme oxygenase 1. J Surg Res 166:e157-164.
Yanamoto H, Mizuta I, Nagata I, Xue JH, Zhang Z, Kikuchi H (2000a) Infarct tolerance accompanied enhanced BDNF-like immunoreactivity in neuronal nuclei. Brain Res 877:331-344.
Yanamoto H, Nagata I, Sakata M, Zhang Z, Tohnai N, Sakai H, Kikuchi H (2000b) Infarct tolerance induced by intra-cerebral infusion of recombinant brain-derived neurotrophic factor. Brain Res 859:240-248.
Yanamoto H, Xue JH, Miyamoto S, Nagata I, Nakano Y, Murao K, Kikuchi H (2004) Spreading depression induces long-lasting brain protection against infarcted lesion development via BDNF gene-dependent mechanism. Brain Res 1019:178-188.
Zhao H, Wang R, Tao Z, Gao L, Yan F, Gao Z, Liu X, Ji X, Luo Y (2014) Ischemic postconditioning relieves cerebral ischemia and reperfusion injury through activating T-LAK cell-originated protein kinase/ protein kinase B pathway in rats. Stroke 45:2417-2424.
Zheng L, Ding J, Wang J, Zhou C, Zhang W (2016) Effects andmechanism of action of inducible nitric oxide synthase on apoptosis in a rat model of cerebral ischemia-reperfusion injury. Anat Rec (Hoboken) 299:246-255.
Zou J, Wang YX, Dou FF, Lü HZ, Ma ZW, Lu PH, Xu XM (2010) Glutamine synthetase downregulation reduces astrocyte protection against glutamate excitotoxicity to neurons. Neurochem Int 56:577-584.
Copyedited by Phillips A, Norman C, Yu J, Li CH, Qiu Y, Song LP, Zhao M
*Correspondence to: Li Cui, Ph.D. or Jia-chun Feng, Ph.D., chuili1967@126.com or fengjcfrank@qq.com.
orcid: 0000-0002-5999-2492 (Jia-chun Feng)
10.4103/1673-5374.194759
Accepted: 2016-10-09
- 中國神經再生研究(英文版)的其它文章
- Nerve growth factor protects against palmitic acidinduced injury in retinal ganglion cells
- Tissue-engineered rhesus monkey nerve grafts for the repair of long ulnar nerve defects: similar outcomes to autologous nerve grafts
- HLA class II alleles and risk for peripheral neuropathy in type 2 diabetes patients
- Rab27a/Slp2-a complex is involved in Schwann cell myelination
- Key genes expressed in different stages of spinal cord ischemia/reperfusion injury
- Methylprednisolone promotes recovery of neurological function after spinal cord injury: association with Wnt/β-catenin signaling pathway activation