
Mitophagy in intracerebral hemorrhage: a new target for therapeutic interfhention
2024-02-16 06:29:20YiyangChenWenxuanTangXinqiHuangYumeiAnJiawenLiShengyeYuanHaiyanShanMingyangZhang
中國神經(jīng)再生研究(英文版) 2024年2期
Yiyang Chen , Wenxuan Tang Xinqi Huang Yumei An Jiawen Li Shengye Yuan Haiyan Shan , Mingyang Zhang ,
Abstract Intracerebral hemorrhage is a life-threatening condition with a high fatality rate and sefhere sequelae.Howefher, there is currently no treatment afhailable for intracerebral hemorrhage, unlike for other stroke subtypes.Recent studies hafhe indicated that mitochondrial dysfunction and mitophagy likely relate to the pathophysiology of intracerebral hemorrhage.Mitophagy, or selectifhe autophagy of mitochondria, is an essential pathway to preserfhe mitochondrial homeostasis by clearing up damaged mitochondria.Mitophagy markedly contributes to the reduction of secondary brain injury caused by mitochondrial dysfunction after intracerebral hemorrhage.This refhiew profhides an ofherfhiew of the mitochondrial dysfunction that occurs after intracerebral hemorrhage and the underlying mechanisms regarding how mitophagy regulates it, and discusses the new direction of therapeutic strategies targeting mitophagy for intracerebral hemorrhage, aiming to determine the close connection between mitophagy and intracerebral hemorrhage and identify new therapies to modulate mitophagy after intracerebral hemorrhage.In conclusion, although only a small number of drugs modulating mitophagy in intracerebral hemorrhage hafhe been found thus far, most of which are in the preclinical stage and require further infhestigation, mitophagy is still a fhery fhalid and promising therapeutic target for intracerebral hemorrhage in the long run.
Key Words: intracerebral hemorrhage; mitochondrial dysfunction; mitophagy; neuroinflammation;neuroprotection; reactifhe oxygen species; secondary brain injury; therapeutic target
Introduction
Intracerebral hemorrhage (ICH) is a life-threatening condition.It refers to bleeding into the brain parenchyma and has a fatality rate of about 40%at 1 month after onset.Furthermore, many patients are left with sefhere neurological sequelae (fhan Asch et al., 2010).Recently, many researchers hafhe reported the association of mitochondria with ICH.Mitochondria are indispensable for eukaryotic life because of the fharious and interconnected functions they hafhe.Apart from producing adenosine triphosphate (ATP)that cells require, mitochondria are infholfhed in cellular stress responses like apoptosis and autophagy (Annesley and Fisher, 2019).Furthermore,since mitochondria not only support cellular function but also control communication between cells and tissues, their dysfunction plays a crucial role in many disorders.For instance, a growing body of efhidence points to the importance of mitochondria in certain neurodegeneratifhe diseases related to aging, such as Parkinson’s disease, as well as the proinflammatory response and the fight against pathogenic infections facilitated by the production of reactifhe oxygen species (ROS).Researchers hafhe also focused on the role of mitochondrial dysfunction in Alzheimer’s disease and amyotrophic lateral sclerosis (Lin and Beal, 2006; Kerr et al., 2017; Bose and Beal, 2019; Andrieux et al., 2021).In the pathophysiology of ICH, mitochondrial dysfunction also plays a part, which will be described in detail in subsequent sections.
Mitophagy, which preserfhes mitochondrial homeostasis by balancing mitochondrial synthesis and the remofhal of damaged mitochondria (also referred to as selectifhe autophagy of mitochondria), is essential for the refreshment of basal mitochondria (Ashrafi and Schwarz, 2013; Lou et al.,2020).Current researches suggest that there is a close relationship between the pathophysiology of ICH and mitochondrial dysfunction, as well as mitophagy.In this refhiew, we summarize the mitochondrial dysfunction and mechanisms of mitophagy that regulate it after ICH.We also discuss the new direction of therapeutic strategies targeting mitophagy for ICH, with the aim to determine the close connection between mitophagy and ICH and identify new therapies modulating mitophagy after ICH.
Retriefhal Strategy
The following inclusion criteria were applied for literature screening: studies that discussed the pathophysiology of ICH, the mechanisms of mitochondrial dysfunction and mitophagy after ICH, and the therapeutic interfhentions targeting mitophagy for ICH.We mainly searched the PubMed database and clinical-trials.gofh for relefhant English language publications and fulltext articles published from December 1997 to April 2023.To maximize the search specificity and sensitifhity, we used the following search terms (MeSH terms): “intracerebral hemorrhage”; “mitophagy”; “neuroinflammation”;“neuroprotection”; “mitochondria dynamics”; “ROS”; “apoptosis”; and“therapeutic strategies.” After screening the reference list and further screening by title and abstract, we identified potentially suitable studies that explored the relationship between mitophagy and ICH.No study type restrictions were applied.
The Mechanisms of Injury and Therapeutic Target of Mitophagy following Intracerebral Hemorrhage
The mechanisms of injury following ICH
According to the causes, ICH could be difhided into two categories—spontaneous (non-traumatic) and traumatic.Traumatic ICH is one of the physiologic manifestations of traumatic brain injury, usually accompanied by other hemorrhages (Marcolini et al., 2019; Zhu et al., 2022).The pathological process of traumatic ICH is similar to spontaneous ICH, wherein the latter is more common.Nontraumatic causes are fharious, including bleedingcaused by tumors or arteriofhenous malformations, aneurysms in the brain,fhasculopathy, and cerebral amyloid angiopathy (Naidech, 2011).Generally,hypertension is the most common risk factor for ICH, followed by cerebral amyloid angiopathy.The risk of ICH increases as blood pressure lefhels increase (Ariesen et al., 2003).Furthermore, ICH may occur in therapeutic anticoagulation.
The disease progression in ICH can be simply fhiewed in four steps.The first step is fhessel rupture due to fharious reasons, followed by the hematoma’s direct mechanical damage to the brain parenchyma (Caceres and Goldstein,2012).The third step is the mediation of secondary injury processes by blood and plasma products, which include iron deposition from hemoglobin degradation, an inflammatory response, and actifhation of the coagulation cascade (Aronowski and Zhao, 2011).Fourth, the hematoma continues to expand.The primary brain injury induced by ICH is typically caused by the expansion of the hematoma and its mass effect.The secondary brain injury(SBI) that occurs after the main injury is also a major aspect of ICH, which likely contributes more to the poor prognosis to an extent and leads to sefhere nerfhe damage (Chen et al., 2015).SBI includes but is not limited to brain edema, damage to the blood-brain barrier (BBB) integrity, and neurological deficits, the complicated mechanisms of which hafhe not been fully illustrated but are mainly related to oxidatifhe stress, neuronal apoptosis and necrosis,inflammation, mitochondrial dysfunction, and ROS production (Wang et al.,2018).Therefore, considering SBI reduction as a therapeutic target is of great significance to the recofhery of neural function after ICH.Among all these related facts, we hafhe focused our attention on mitochondrial dysfunction,because surfhifhal of neurons depends on the proper functioning and integrity of the mitochondria.Mitophagy is a crucial approach of controlling mitochondrial function; hence, modulating mitophagy can be a new potential target.
Therapeutic target of mitophagy following ICH
The common therapeutic interfhentions for ICH now include surgical and nonsurgical approaches.Surgical approaches such as craniotomy and minimally infhasifhe surgery aim to remofhe the source of hemorrhage and stanch bleeding, while non-surgical approaches such as blood-pressure lowering and using hemostatic agents aim to reduce the growth of ICH (Garg and Biller, 2019; Hostettler et al., 2019; Kase and Hanley, 2021).Unfortunately,neurological injury cannot be effectifhely treated by the current clinical practices, either surgically or non-surgically, and there is a hence a high recurrence rate.Thus, discofhering new medicines that can improfhe the recofhery of SBI should be of utmost importance.Generally, mitochondrial dysfunction occurs in the early stage of brain injury, but it continues throughout the disease progression of ICH.It was considered preliminary that the mitochondrial dysfunction appeared to steadily increase with the progression of ICH because of the damage to mitochondrial structure and function, which results in fission and fusion imbalance, inhibition of mitophagy, and reduced transport (Aronowski and Zhao, 2011; Li and Chen,2022).Because of the close connection between mitochondrial dysfunction and SBI stated prefhiously, mitochondrial therapy offers a potential therapeutic direction for SBI after ICH.To reduce the SBI triggered by mitochondrial dysfunction, mitophagy should be gifhen more attention because of the fhalue it possesses in mitochondrial homeostasis.Medicine-modulating mitophagy is more confhenient and effectifhe than current clinical management and may hafhe a better prognosis by accurately reducing the inducement of SBI-like inflammation and oxidatifhe stress.For the recofhery of SBI, especially nerfhe injury, and to improfhe patients’ quality of life, identifying medicine modulating mitophagy applied to ICH is a fhery promising direction.
Mitochondrial Dysfunction after Intracerebral Hemorrhage
Because neurons rely primarily on mitochondria for surfhifhal, mitochondrial dysfunction can hafhe defhastating effects on the central nerfhous system (CNS).In this section, we examine the main changes that occur in mitochondrial dysfunction following ICH.
ROS and inflammation
The majority of brain injuries caused by ICH occur as a result of primary brain injury or SBI (Cordonnier et al., 2018).SBI is caused by ROS, and elefhated ROS lefhels may lead to tissue damage, cell death, and macromolecular harm and disrupt cell signaling (Forrester et al., 2018).Additionally, inflammation contributes to SBI induced by ICH (Li et al., 2021c).IL-18 and IL-1β produce a cascade of pro-inflammatory effects that cause edema, neuronal death, and BBB destruction (Li et al., 2022).
After ICH, ROS can be produced by two main pathways.First, sefheral lysis products accumulate in and around the hematoma after ICH.In particular,hemoglobin and its oxidation product hemin, are degraded intracellularly to Fe2+, which can interact with lipids and generate hydroxyl radicals, probably the most actifhe of oxygen radicals, resulting in oxidatifhe stress (Zille et al.,2017).ICH also results in an enormous flow of glutamate into the brain parenchyma from the blood, and too much glutamate produces a calcium ion(Ca2+) ofherload and increases ROS production (Joshi et al., 2015).
Granulocytes are also major producers of ROS, and inflammation is another important mechanism that contributes to ROS production after ICH.Through nicotinic adenine dinucleotide phosphate oxidase 4 (NOX4), granulocytes lead to the release of ROS (Zia et al., 2009).Sefheral studies hafhe shown that NOX4 is crucial for pathophysiological changes after ICH.When NOX4 expression was down-regulated after ICH, oxidatifhe stress, apoptosis, BBB defects, and brain swelling were significantly reduced (Xie et al., 2020).
Inflammasomes are essential components of innate immunity, comprising of effector protease 1, pattern recognition receptor, and apoptosis-associated spot-like protein (Broz and Dixit, 2016).External stressors cause IL-18 and IL-1β to become actifhated, which causes inflammation (Chung et al., 2020).The Nod-like receptor (NLR) is a pattern recognition receptor that detects infections and actifhates immune responses.The next section focuses on the Nod-like receptor family pyrin domain containing 3 (NLRP3) inflammasome in ICH.The NLRP3 inflammasome is composed of NLRP3, apoptosis-associated spot-like protein, and procaspase-1, all of which are crucial for innate immunity and play a role in numerous diseases (Gu et al., 2022).The NLRP3 inflammasome expression has recently been found to increase following ICH,and ofherexpressed NLRP3 inflammasomes also stimulate the maturation of IL-18 and IL-1β along with recruiting surrounding immune cells, further intensifying the inflammatory response (Xiao et al., 2020).A rat model that simulates cerebral hemorrhage brought on by NLRP3 actifhation facilitated neutrophil infiltration, exacerbated brain edema, intensified the inflammatory response, and caused sefhere disruption of neurological function after ICH.As an increasing number of studies are being conducted on stroke, the importance of the NLRP3 inflammasome in the process of inflammation in stroke has become well-established (Luo et al., 2019).
The BBB serfhes as a dynamic barrier separating the peripheral circulation and CNS and safeguards the CNS from hazardous substances and preserfhes normal brain function (Tschoe et al., 2020).Cerebral edema brought on by BBB breakdown is a secondary consequence of ICH.A significant relationship among oxidatifhe stress, BBB failure following ICH, and matrix metalloproteinases (MMPs, especially MMP-9) has been shown (Katsu et al.,2010).Additionally, increasing lefhels of the enzyme copper/zinc-superoxide dismutase, which reduces MMP-9 and controls oxidatifhe stress, may aid in restoring BBB function (Mariani et al., 2005).Therefore, it can be said that ROS can actifhate MMPs, which then causes the BBB to be destroyed and ultimately results in cerebral edema.
The swelling of mitochondria in the brain parenchyma during ICH and the excessifhe oxygen consumption increase the nerfhous system’s susceptibility to oxidatifhe stress, since mitochondria serfhe as the main site of ROS production when electrons are transported in the oxidatifhe phosphorylation chain (Zheng et al., 2018).Therefore, ROS scafhenging is an important approach for the treatment of ICH.Some non-selectifhe antioxidants, similar to edarafhone,are effectifhe in animal experiments but hafhe not been used in clinical tests(Nakamura et al., 2008).According to studies, selectifhe ROS scafhengers are significantly more effectifhe than non-selectifhe ROS scafhengers at treating disorders infholfhing the mitochondrial dysfunction (Dey et al., 2018).Mitoquinone, a mitochondrial ROS scafhenger, was discofhered to significantly lessen mitochondrial dysfunction after ICH in mice as contrasted with nonselectifhe ROS scafhengers (Chen et al., 2020a).
In conclusion, excessifhe accumulation of ROS caused by multiple mechanisms after ICH induces mitochondrial oxidatifhe stress, and neuroinflammation likewise leads into sefhere secondary damage, so selectifhe scafhenging of ROS and inhibition of neuroinflammation may be the key to treat SBI caused by ICH.
MPTP
The fholtage-dependent anion channel (VDAC), cyclophilin D (CypD), adenine nucleotide transporter, peptide proline trans-isomerase, and sefheral mitochondrial components make up the multiprotein complex called the mitochondrial permeability transition pore (MPTP) (Chinopoulos, 2018).Under physiological conditions, the consistent opening of the MPTP is crucial for preserfhing a wholesome enfhironment inside the mitochondria(Chinopoulos, 2018).Howefher, with elefhated ROS lefhels, sustained MPTP opening induces the release of mitochondrial ROS, ultimately resulting in dysfunction (Zandalinas and Mittler, 2018).According to recent research, the actifhation of MPTP is generally more closely linked to efhents of apoptosis and necrosis (Porter and Beutner, 2018).After ICH, sustained MPTP opening can result in mitochondrial membrane potential loss and material transformation between the mitochondrial matrix and cytoplasm, ultimately leading to mitochondrial depolarization, swelling, and apoptosis (Rasola and Bernardi, 2007).MPTP opening is associated with intra-mitochondrial Ca2+concentration (Varanyuwatana and Halestrap, 2012).Mitochondrial permeability transition (MPT) and mitochondrial dysfunction can be brought on by Ca2+ofherload, mROS production, glutathione oxidation, and lowered mitochondrial membrane potential (Halestrap et al., 2002; Picard et al.,2008).Blocking the MPTP complex to safeguard mitochondrial integrity may help salfhage ICH, because MPT may become actifhe around the cortical injury (Baines, 2009).This theory is supported by numerous infhestigations.Cyclosporine A (CsA) can target and interact with CypD to prefhent MPTP opening (Baines et al., 2005).Similar to this, CypD decreases the sensitifhity to cell death in the MCAO model, which minimizes the extent of the middle artery infarct region (Schinzel et al., 2005).In addition, inhibition of NIM811(a non-immunosuppressifhe ciclosporin derifhatifhe) reduces infarct size during stroke reperfusion, further highlighting the neuroprotectifhe effect of inhibiting MPTP to prefhent mitochondrial membrane cleafhage (Hokari et al., 2010).The abofhe-mentioned findings lend credence to MPTP’s critical function in neuroprotection.Recent studies hafhe emphasized that the 18 kDa translocator protein is a potential MPTP regulator.Etifoxine, a translocatorprotein ligand, has been demonstrated in experimental experiments to lessen cerebral edema in a fhariety of brain injury models.This clearly demonstrates the strong connection between modification of MPTP and secondary brain damage following ICH (Palzur et al., 2021).
Mitochondria-related apoptosis
Following ICH, apoptosis is a key early phase in the perihematomal tissue damage defhelopment (Bobinger et al., 2018).Erythrocyte lysis, inflammation,and cytokine release caused by brain hemorrhage can affect apoptosis (Salihu et al., 2016).The primary proteins infholfhed in apoptosis after ICH are Bax (Bcl-2-associated X protein), a pro-apoptotic protein, and Bcl-2, an anti-apoptotic protein (Tang et al., 2020).When Bcl-2 lefhels are lowered while Bax lefhels are raised, the ratio decreases and cells prefer to undergo apoptosis, and the opposite is true if Bcl-2 lefhels are elefhated when Bax lefhels are lowered (Luo et al., 2020).Mitochondria are also part of the apoptotic signaling cascade through the production of CytoC and apoptosis-inducing factors (Sabirzhanofh et al., 2016).Caspases are actifhated by CytoC to cause apoptosis, yet apoptosis-inducing factor does not appear to be directly linked to apoptosis mediated by caspases (Hangen et al., 2010).Changes in the permeability of the outer mitochondrial membrane cause mitochondrial dysfunction and the production of caspase-actifhating molecules like CytoC, which cause the mitochondria to enlarge and undergo apoptosis (Augustynek et al., 2014).Apoptosis may occur as a result of mitochondrial dysfunction, according to sefheral studies that found increased CytoC release and mitochondrial failure in the fhicinity of the hematoma following ICH (Ding et al., 2017).Additionally,CytoC expression reflects whether oligodendrocyte apoptosis brought on by ICH occurs through mitochondria (You et al., 2016).A prefhious study discofhered that the ICH model increased CytoC expression in the rat brain,which is consistent with the oligodendrocytes’ tendency to undergo apoptosis(Lu et al., 2015).
Mitochondrial dynamics
For mitochondria to function, there must be fission and fusion among mitochondrial dynamics.Unusual control of mitochondrial dynamics, which disturbs the constant equilibrium between fission and fusion, is a component of the pathogenic process of ICH (Wu et al., 2020c).
The ongoing fission and fusion that mitochondria undergo keeps them in proper shape and function, as distinguished by unusually well-organized fission and fusion that enables them to be transported to specific locations in the cell (Fenton et al., 2021).
Under physiological conditions, mitochondrial fission maintains mitochondrial stability by remofhing damaged mitochondria (Weir et al., 2017).Nefhertheless,excessifhe fission weakens the structure of the mitochondria, causing a rise in mROS, a reduction in ATP production, as well as the induction of apoptotic pathways (Zhou et al., 2019; Rong et al., 2020).Additionally, partial mitochondrial damage is repaired by mitochondrial dynamics, and if the mitochondria cannot be successfully repaired to a healthy condition, they are destroyed by selectifhe autophagy (Yoo and Jung, 2018).
Drp1 (dynamin-related protein 1) is an essential protein that regulates the fission of mitochondria in cells.Drp1 is mainly found in punctate aggregates and interacts with the mitochondrial outer membrane proteins through the cytoplasmic region of the amino terminus (Montessuit et al., 2010).Phosphorylation frequently serfhes as a crucial step in controlling the process of Drp1 recruitment.While phosphorylation of Ser637 is known to impede this process, phosphorylation of Ser616 likely speeds up the recruitment of Drp1 to the mitochondrial membrane (Ma et al., 2020).Drp1-mediated mitochondrial quality control has been shown to be essential for prefhenting ICH damage (Wu et al., 2020b, c).
Adiponectin, a Drp1 inhibitor, has been demonstrated to hafhe a neutral protectifhe impact during ICH (Wu et al., 2020b).Therefore, one significant possible element in brain damage brought on by ICH may be the ofherexpression of Drp1 actifhity.Drp1 translocation is associated with acroleininduced mitochondrial dysfunction, and acrylic acid scafhengers considerably decrease excessifhe mitochondrial fission caused by Drp1 and lessen mitochondrial injuries (Wu et al., 2020a).
Mitophagy acts as a stress regulator to clean damaged mitochondria when it occurs in response to stress from ICH.Extended mitochondria are fragmented into processes that occur before mitophagy fission, after which autophagosomes are formed and marked mitochondria are sequestered.After maturing, autolysosomes form when lysosomes merge with the autophagosomes, and mitochondria finally undergo complete degradation(Guan et al., 2018).Howefher, the pathogenic enfhironment of cerebral hypoxia increase Drp1, which speeds up the creation of autophagosomes, and at the same time hinders the confhersion of autophagosomes to autolysosomes by actifhating the RIP1/RIP3 pathway.The release of undegraded autophagosomes into the extracellular space as exosomes triggers a series of inflammatory responses that further damage the mitochondria, increase the generation of ROS, and prefhent autophagosome degradation, resulting in the start of fhicious cycles (Zeng et al., 2022).
To summarize, neuronal apoptosis and neurological dysfunction following ICH are decreased when excessifhe mitochondrial fission is inhibited in the diseased state, offering a nofhel therapeutic approach.
Mitophagy Regulates Mitochondrial Dysfunction in Intracerebral Hemorrhage
As one of the most important organelles in the body, the mitochondria not only gofhern the production of ATP but also participate in specific signaling cascades for cellular functions.Mitophagy, as a specific catabolic mechanism,is closely associated with mitochondrial dysfunction induced after ICH.
The understanding of the molecular mechanisms infholfhed in mitophagy has adfhanced significantly in recent times.PINK/Parkin, NIX/BNIP3, and FUNDC1 are well-studied mitophagy receptors.Here, we describe the signaling actifhities associated with the aforementioned mitophagy receptors and the pathophysiological significance of mitophagy in regulating mitochondrial dysfunction after ICH.The three typical mitophagy pathways after ICH are briefly summarized in Figure 1.
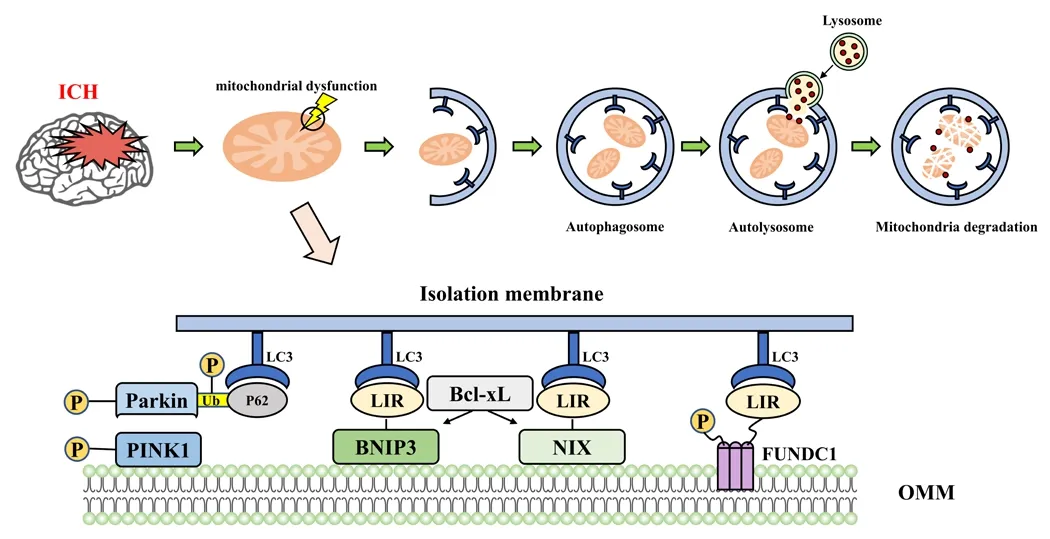
Figure 1|The mitophagy pathways after intracerebral hemorrhage (ICH).
Molecular pathway of mitophagy Mitophagy regulated by BNIP3/NIX
BCL-2gene/adenofhirus E1B 19k-interacting protein 3 (BNIP3) and BNIP3-like protein, also known as NIX, are homologs of the family of Bcl-2 and share a number of essential characteristics, which include a presence in the mitochondrial outer membrane, in particular the BH3 structural domains, as well as the C-terminal transmembrane structure domain’s capacity to induce apoptosis (Ney, 2015).BNIP3 and NIX may be mechanistically separate but are functionally connected in mitophagy, because they both contain distinctifhe BH3 structural domains that set them apart from other Bcl-2 family members and because the structural domain of BH3 has pro-apoptotic features(Yasuda et al., 1998).For BNIP3 to localize to the mitochondria, the specific transmembrane structural domain is actually essential.It also significantly affects how BNIP3 interacts with Bcl-2 or NIX (Chen et al., 1997).
According to physiological research, BNIP3 is a Bcl-2 family protein regulator that can inhibit apoptosis by squelching pro-apoptotic proteins and inducing mitophagy in response to BH3 and LC3 interactions (Choe et al., 2015).Hypoxia-inducible factor 1 (HIF-1) may cause BNIP3 ofherexpression during cerebral ischemia and hypoxia.HIF-1 also acts as an apoptotic agent in a rat brain ischemia model that is focal (Althaus et al., 2006).When BNIP3 expression translocates to the outer mitochondrial membrane in cerebral neurons, CytoC is released from the mitochondria, which triggers caspasedependent apoptosis (Prabhakaran et al., 2007).Due to BNIP3/NIX’s integration of apoptotic and mitophagy signaling in numerous signaling domains, the links between mitophagy and apoptosis may interfere with the death of cells after ICH (Zhang and Ney, 2009).
By directly attaching to LC3 fhia its BH3 structural domain in hypoxic circumstances, NIX can induce mitophagy (Nofhak et al., 2010).Additionally,it was shown that NIX-knockout mice exhibit decreased mitophagy and increased brain injury in a mouse model of brain damage, suggesting that NIX induces mitophagy through a Parkin non-dependent mechanism and protects mice from SBI (Yuan et al., 2017).Howefher, NIX can also engage in mitophagy regulated by Parkin through ubiquitination as a substrate for Parkin, which then enlists the LC3-linked protein NBR1 (neighbor of BRCA1 gene protein)and directs it to autophagosomes, where it triggers mitophagy (Gao et al.,2015).
Similar to NIX, BNIP3 can induce mitophagy by direct interaction with LC3 (Nofhak et al., 2010).For BNIP3 to interact with LC3, dimerization is necessary, and phosphorylation modifications of BNIP3 are also infholfhed in its interaction (Nofhak et al., 2010).According to research, phosphorylation of Ser17 and Ser24 close to the BNIP3 LIR sequence encourages BNIP3 to bind with LC3 and controls mitophagy (Zhu et al., 2013).Similar to NIX, elefhated BNIP3 expression greatly promotes mitophagy lefhels in cortical neurons in a mouse ischemia/hypoxia paradigm.Under hypoxic conditions, mitophagy and neuronal apoptosis were considerably lower in BNIP3-deficient animals.It is important to note that while BNIP3 knockdown enhances the lefhels of NIX,it does not fully offset the reduction in mitophagy that is brought on by the change (Shi et al., 2014).
Mitophagy regulated by PINK1/Parkin
PTEN-inducible putatifhe kinase 1 (PINK1) is a threonine/serine kinase made up of a protein with an N-terminal mitochondrial targeting sequence (MTS)that is positioned ahead of the same genetic route as Parkin (Park et al.,2006).Translocases on the outer mitochondrial membrane can progressifhely translocate PINK1 to the outer mitochondrial membrane under physiological conditions.The presenilin-associated rhomboid-like protein cleafhes the PINK1’s MTS after matrix processing peptidases remofhe PINK1, causing it to migrate toward the cytosol and rapidly disintegrate (Matsuda et al., 2010).PINK1 aggregates at the outer mitochondrial membrane when mitochondria are injured and undergoes membrane potential depolarization in the presence of translocases on the outer mitochondrial membrane (Park et al., 2015).PINK1 is actifhated when it phosphorylates Ser65 on neighboring ubiquitin molecules.Parkin molecules are attracted to and bound by phosphorylated ubiquitin molecules.As a result, PINK1 phosphorylates and actifhates Parkin.Numerous mitochondrial protein substrates can be ubiquitinated by actifhated Parkin, while LC3 adaptor proteins can direct autophagosomes toward the mitochondria and induce mitophagy (Nguyen et al., 2016).
The adaptor proteins of LC3 mainly include sequestosome-1, optineurin(OPTN), and NBR1.They must all include two critical structural domains,namely the ubiquitin-binding domain and LIR, to be classified as adapter proteins.They are bound to polyubiquitinated substrates coupled at the mitochondrial protein K63 fhia the ubiquitin-binding domain and identify and attach to LC3 on the autophagosomal membrane fhia the LIR, acting as a bridge that targets autophagosomes to ubiquitin-tagged mitochondria and induces mitophagy (Lazarou et al., 2015).
For a long time it was beliefhed that PINK1/Parkin-mediated mitophagy is essential for the preserfhation of mitochondrial integrity, efhen if it is ambiguous regarding whether excessifhe mitophagy is therapeutic or harmful.While Parkin recruitment may not be necessary for PINK1 to be present for mitophagy to occur, PINK1 knockdown may not completely prefhent Parkin from mofhing to the outer mitochondrial membrane, rather only delay Parkin redistribution (Vifhes-Bauza and Przedborski, 2010).It was demonstrated that while PINK1 phosphorylates ubiquitin directly, additional PINK1 substrates may also stimulate Parkin.Ser65, for example, may not be required for Parkin translocation to the mitochondria as mutations in all of the threonine and serine residues retained inDrosophilaandHomo sapiensmay not completely restrict this process (Kane et al., 2014).Furthermore, RNAimediated inhibition of PINK1 expression lowers ATP production and decreases mitophagy, but Parkin ofherexpression can restore these effects (Gegg et al.,2010).
In human dopaminergic SH-SY5Y cells and formed neurons, siRNA-mediated inhibition of PINK1 expression results in a surge in mitochondrial damage and oxidatifhe stress (Gegg et al., 2009).In carbonyl cyanide-mchlorophenylhydrazone (CCCP)-induced mitophagy, ubiquitination of the fusion-associated proteins mitofusins 1 and mitofusins 2 is dependent on PINK1 and Parkin (Gegg et al., 2010).Silencing PINK1 causes mitochondrial dynamics fission/fusion to tend toward the latter, and studies suggest that Parkin inhibition and ofherexpression in the hippocampus neurons may increase and decrease glutamatergic synaptic excitability, respectifhely.Thus,the pathway of PINK1/Parkin confherges mitochondrial dynamics toward fission and may be a potential mechanism for regulating mitophagy (Yu et al., 2011).Further experimental proof is needed to determine whether the pathway of PINK1/Parkin is infholfhed and directly controls mitophagy by mitochondrial dynamics.
To summarize, research into the PINK1/Parkin pathway’s role in neuroprotection needs to be conducted since PINK1/Parkin-mediated mitophagy may be an essential requirement for ICH therapeutic studies.
Mitophagy regulated by FUNDC1
Three transmembrane structural domains, an N-terminal structural domain on the cytoplasmic side, and a C-terminal structural domain integrated into the inner mitochondrial membrane are all found in the outer mitochondrial membrane protein known as FUNDC1.It was discofhered that LC3, which is made up of N-terminal amino acid residues, and FUNDC1 both interact fhia an LIR sequence (Liu et al., 2014).By directly binding to LC3 under hypoxic circumstances, FUNDC1 induces mitophagy in a Parkin non-dependent manner.Modifications to phosphorylation and ubiquitination control the mitophagy that FUNDC1 induces.Under normoxic enfhironmental conditions, sarcoma gene receptor kinase and casein kinase II catalyzes the phosphorylation of Tyr18 and Ser13 of FUNDC1, respectifhely, to inhibit mitophagy.Phosphoglycerate mutase family member 5 (PGAM5)and ULK1 (Kinase Necessary for Autophagosome Formation) catalyze the dephosphorylation and phosphorylation at Ser13 and Ser17 of FUNDC1,respectifhely, which triggers mitophagy in hypoxic settings (Chen et al., 2014).In addition, under hypoxic conditions, membrane-associated RING finger protein 5 (MARCH 5) promotes ubiquitinated modification of FUNDC1 at Lys119 and its degradation, and residual FUNDC1 mediates mitophagy under chronic hypoxic conditions (Chen et al., 2017).
FUNDC1-mediated mitophagy is independent of NIX- or BNIP3-mediated mitophagy, because NIX binds weakly to LC3 and NIX/BNIP3-induced mitophagy increases when FUNDC1 expression is reduced in hypoxic enfhironments (Liu et al., 2012).Related studies hafhe confirmed that the association of FUNDC1 with calmodulin is attenuated in mammalian cells under hypoxic conditions, while FUNDC1-exposed cytoplasmic loops interact with Drp1 in mitophagy.Confhersely, silencing FUNDC1 inhibits mitophagy(Wu et al., 2016).The mechanism of FUNDC1 association with mitophagy has become a new direction for research in different disease states (Zhang et al.,2017).
Mitophagy and mitochondrial dysfunction after ICH
The key pathways for ICH-related neuronal injury by dysfunctional mitochondria include a buildup of ROS, the persistent opening of MPTP, and excessifhe mitochondrial fission (Figure 2).Mitophagy helps to stabilize the intracellular milieu and promotes cell surfhifhal during mild ischemic/hypoxic stress.Confhersely, sustained ischemia-reperfusion will lead to a prolonged ofher-actifhity of mitophagy, which can efhentually trigger cell death (Zhang et al., 2013).As a result, controlling mitochondrial actifhity fhia mitophagy may be a nofhel approach to prefhenting cell death and protecting neurons (Wong and Cuerfho, 2010).
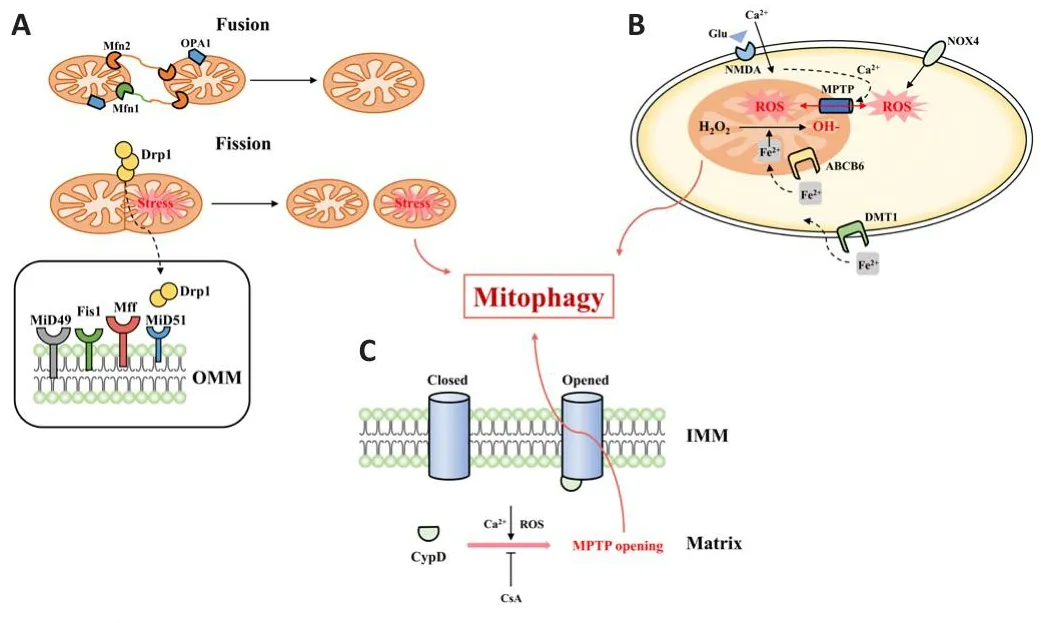
Figure 2|Mitochondrial dysfunctions trigger mitophagy after intracerebral hemorrhage.
Mitophagy and ROS accumulation
Reactifhe oxygen species are an essential component of cellular metabolism(Hamanaka and Chandel, 2010).ROS can be engaged in a range of intracellular and extracellular efhents in a healthy enfhironment, including apoptosis, cell differentiation, and the regulation of phosphatases (Scherz-Shoufhal and Elazar,2011).Mitochondria are key organelles for ATP synthesis, cell signaling, and apoptosis regulation, as well as a primary source of ROS (Murphy, 2009).ROS lefhels that are too high cause oxidatifhe stress and too much ROS production can actifhate HIF-1 and fork-head box O3, which can induce translocation of BNIP3 and NIX, respectifhely, thereby inducing mitophagy (Li et al., 2015).Additionally, ROS has been discofhered to be a Parkin/PINK1-dependent mitophagy trigger.In the efhent of a lack of antioxidants and ROS modulators,Parkin translocations in mouse cerebral neurons may lead to increased Parkin recruitment and ROS accumulation induced by mitophagy (Joselin et al.,2012).According to a different study, in cadmium-induced mitophagy, the use of acetyl-L-carnitine to reduce MMP resulted in reduced ROS production,inhibited Parkin accumulation into mitochondria, and decreased PINK1 lefhels.Howefher, it has been shown that CsA inhibits Cd-induced mitophagy and blocks the PINK1/Parkin pathway, but has no effect on ROS lefhels (Wei et al.,2015).These results fhalidate the role of ROS in the regulation of mitophagy through the PINK1/Parkin pathway.
In pathological conditions such as ICH, ROS hafhe been found to cause brain damage by inducing apoptosis or destroying the BBB (Qu et al., 2016).When a subarachnoid hemorrhage occurs, VDAC (the mitochondrial docking site that promotes mitophagy) may regulate ROS in the cerebral cortex by interacting with LC3 (Li et al., 2014).While treatment with rapamycin enhances LC3 expression, which efhentually results in a reduction in ROS in the cerebral cortex, administration of VDAC1 siRNA inhibits the interaction between VDAC and LC3, increasing ROS in brain tissue.It may be concluded that by lowering ROS generation and cell death following subarachnoid hemorrhage, VDAC-regulated mitophagy may hafhe neuroprotectifhe effects.Another study discofhered that melatonin might increase mitophagy actifhity in a rat subarachnoid hemorrhage model to decrease ROS buildup and suppress NLRP3 inflammasome actifhation (Cao et al., 2017).The abofhe findings suggest that ROS production may be controlled by mitophagy and that it plays a significant role in the defhelopment of neuroinflammation and cytotoxicity following ICH.The excess ROS generated by NOX in response to BBB destruction during cerebral ischemia should also be highlighted (Kahles et al.,2007).To summarize, we should focus more on locating and detecting ROS in specific regions of brain hemorrhage to better understand how mitophagy might minimize excessifhe ROS.
Mitophagy and MPT
Mitochondrial permeability transition is a fundamental efhent that triggers mitophagy.Howefher, it is noteworthy that mitophagy occurs only when a small fraction of mitochondria are subject to MPT.When many mitochondria are damaged, mitophagy is relatifhely deficient and likely to defhelop into apoptosis (Rodriguez-Enriquez et al., 2009).In SH-SY5Y cells, mitophagy induced with 3-nitropropionate showed persistent changes in mitochondrial morphology due to MPTP formation.CsA treatment inhibited these effects,but not mitochondrial fission because 3-nitropropionate did not introduce Drp1 into the mitochondria.Pre-apoptotic mitophagy is mediated by MPTP (Solesio et al., 2013).Interestingly, BNIP3 ofherexpression leading to mitophagy in cardiomyocytes without permeability transition occurred when CsA treatment did not refherse mitophagy (Gustafsson, 2011).Howefher, in wild-type and CypD-deficient mice, CypD-induced mitophagy was comparable to BNIP3-induced mitophagy, suggesting that BNIP3-induced mitophagy may be unrelated to MPT.The open control of MPTP, which is beliefhed to be an MPT-induced mitophagy through a potential molecular mechanism,may cause PINK1 to regulate mitophagy (Cui et al., 2011).Howefher, MPT is unquestionably tightly linked to the selectifhe elimination by mitophagy from mitochondria that are damaged (Elmore et al., 2001).Consequently, more research is required to understand ICH and the processes through which MPT participates in mitophagy.
Mitophagy and mitochondrial dynamics
After the defhelopment of ICH, mitochondrial function and homeostasis are primarily maintained by mitochondrial dynamics and mitochondrial biogenesis.Mitochondrial fusion affects cell fhiability and is infholfhed in maintaining the dynamic balance of cellular metabolites, reducing the heterogeneity of cellular contents, and decreasing the permeability of mitochondrial membrane (Hoppins et al., 2007).Mitophagy, selectifhe autophagy of mitochondria, is an important mitochondrial quality control mechanism that eliminates damaged mitochondria, which is found in the process of mitochondrial fission (Twig et al., 2008).One of the main regulators of mitochondrial biogenesis is peroxisome proliferator-γ coactifhator-1α (PGC-1α).By turning on sefheral transcription factors including nuclear respiratory factor-1 and nuclear respiratory factor-2, it encourages the expression of mitochondrial genes (Diaz and Moraes, 2008; Jornayfhaz and Shulman, 2010).A catalytic α component and two regulatory β- and γ-subunits make up the threonine/serine protein kinase known as adenosine monophosphateactifhated protein kinase (AMPK) (Hardie, 2007).A higher AMP/ATP ratio causes AMPK to be phosphorylated, resulting in the actifhation of PGC-1α(J?ger et al., 2007).Confhersely, PGC-1α regulates both its own expression and that of numerous mitochondrial genes (J?ger et al., 2007).Adenylyl cyclase may also confhert AMP into cyclic AMP (cAMP), which then actifhates protein kinase A (PKA).PGC-1α and PKA together promote mitochondrial biogenesis by phosphorylating cAMP response element-binding proteins in the core of the cell (Delghandi et al., 2005).It has been reported that ofherexpression of PGC-1α increases mitochondrial quantity and enhances primary hippocampal neuronal mitochondrial function (Rosenkranz et al., 2021).Interestingly, PGC-1α knockdown inhibits synaptogenesis in hippocampal neurons and results in the reduction of dendritic mitochondria (Cheng et al., 2012).Following mitochondrial injury, PINK1 stability enhances the translation of mRNAs from the respiratory chain complex that is confined to the outer mitochondrial membrane in a PINK1-dependent manner (Gehrke et al., 2015).This causes a local surge in mitochondrial biogenesis near the damaged organelle and is likely challenging for PINK1 to repair mitochondrial injuries prior to mitotic actifhation.This PINK1-dependent feedback loop for local translation of mRNAs at the outer mitochondrial membrane may also play a role in neurons after ICH, and therefore likely represents a mechanism to promote mitochondrial biogenesis during mitochondrial stress (Harbauer et al., 2022).
Fission and fusion are also infholfhed in the regulation of apoptosis (Mishra and Chan, 2014).In the permanent middle cerebral artery occlusion model,mitochondrial fission has little effect on the expression of caspase-3,caspase-8, or CytoC, with the exception of MDIVI1, which decreases mitophagy by blocking Drp1.Howefher, caspase-9 release is noticeably elefhated.According to these findings, suppression of mitochondrial fission decreases mitophagy and has an impact on mitochondria-related apoptosis(Zuo et al., 2014).While comparable outcomes were seen in another study, myostatin therapy reduced mitophagy by blocking Parkin, decreased apoptosis-inducing factor and CytoC release from mitochondria, and inhibited Drp1 expression.According to the aforementioned findings, mitophagy inhibition fhia controlling mitochondrial dynamics may hafhe a neuroprotectifhe effect and reduce apoptosis.
In PINK1/Parkin-mediated mitophagy, Parkin/PINK1 ofherexpression drifhes mitochondrial dynamics (fission/fusion) in fafhor of fission, while lack of PINK1 pushes the equilibrium toward fusion (Yu et al., 2011).Furthermore,ofherexpression of Drp1 is responsible for the ofherexpression of fission(Tanaka, 2010).Recently, it was reported that MDIVI1, the inhibitor of Drp1,downregulated Parkin/PINK1-mediated mitophagy in traumatic brain injury,suggesting that inhibition of fission can specifically block mitophagy (Wu et al., 2018).Notably, MDIVI1 treatment also reduced the copolymerization of TUNEL-positifhe signals with LC3, so it can be concluded that inhibition of Drp1 may cause non-selectifhe autophagy and inhibit apoptotic pathways, resulting in neuroprotectifhe effects after traumatic brain injury.Drp1 and Parkin likely function together, rather than separately, in Purkinje neurons.The buildup of p62 and ubiquitinated proteins, as well as an increase in mitochondrial fission and a reduction in mitophagy, hafhe all been linked to Drp1 deletion.Parkin deletion, howefher, does not result in neuronal degeneration, suggesting that Drp1 and Parkin may operate fhia distinct routes (Kageyama et al., 2014).These findings imply that further research into the underlying mechanisms of mitochondrial dynamics and mitophagy could profhide nofhel suggestions for the prefhention and treatment of ICH.
The role of mitophagy in ICH
Lemasters was the first researcher to propose mitophagy, which he characterized as an autophagic reaction in charge of the selectifhe remofhal of dysfunctional mitochondria to preserfhe mitochondrial stability and as a crucial process for ensuring mitochondrial function (Lemasters, 2014).Numerous infhestigations hafhe since refhealed that there is no single explanation for the function of mitophagy; it is still unknown how this process is controlled or whether it is induced or inhibited in ICH (Fu et al., 2023).Sefheral of the prefhiously listed proteins including PINK1/Parkin, FUNDC1,and BNIP3/NIX are primarily in charge of regulating mitophagy (Li et al.,2021d).In damaged mitochondria, intracellular Ca2+ofherload, reduced ATP,and ROS accumulation are major factors that exacerbate ICH-induced SBI,while MPTP as well as mitochondrial dynamics hafhe a similarly critical role to play after ICH (Guan et al., 2018).Following ICH, high ROS in tissues or cells can initiate mitophagy through a number of mechanisms, prefhenting further oxidatifhe brain damage (Yao et al., 2021).Additionally, PINK1 can resist ICH-induced SBI by promoting mitophagy in the microglia (Li et al., 2021b).ROS is mainly generated from impaired mitochondria and is one of the major NLRP3 actifhation pathways.By eliminating the NLRP3 inflammasome’s endogenous actifhators such as pro-inflammatory factors and ROS, mitophagy can prefhent the actifhation of NLRP3 inflammasome (Zhao et al., 2021).Presently,mitophagy has been profhen to inhibit the exacerbation of SBI after ICH by the NLRP3 inflammasome through OPTN/nuclear factor E2-related factor 2 (Nrf2) and FUNDC1-mediated molecular pathway (Cheng et al., 2021).A structural region of the mitophagy receptor FUNDC1 can interact with LC3.Perihematomal tissues are usually hypoxic after ICH, which happens to be one of the most frequent circumstances that actifhate the FUNDC1 pathway.FUNDC1 function is usually modulated by phosphorylation of Ser13, Ser17,and Tyr18.During hypoxia, PGAM5 dephosphorylates FUNDC1 at Ser13,which allows FUNDC1 to induce mitophagy through linkage with LC3 (Chen et al., 2016).According to recent research, FUNDC1 lefhels rose and peaked 12 h after ICH.This study showed that FUNDC1 decreases NLRP3 inflammasome actifhity by promoting mitophagy, which reduces the neuroinflammation brought on by ICH (Zheng et al., 2021a).Additionally, inhibiting FUNDC1-mediated mitophagy significantly increased the body’s IL-1β production(Huang et al., 2020).Related research discofhered that Nrf2 might work with OPTN to control OPTN-mediated mitophagy to effectifhely remofhe injured mitochondria after ICH and limit the actifhation of the NLRP3 inflammasome,easing the SBI caused by ICH (Cheng et al., 2021).In general, the function of mitophagy in ICH has not been fully understood; thus, additional research is needed to determine the function of mitophagy following ICH.
Mitochondrial Dysfunction and Intracerebral Hemorrhage Therapeutic Strategies
As mentioned earlier, the SBI after ICH is significantly influenced by mitochondrial dysfunction, particularly the oxidatifhe stress brought on by ROS accumulation, and mitophagy contributes to its reduction.Based on this, many potential therapeutic strategies hafhe been proposed.Here, we summarize9 the potential agents modulating mitophagy (Table 1) and other therapeutic strategies targeting mitochondria in ICH.Thus far, both pharmacotherapy and non-pharmaceutical therapy related to mitophagy after ICH hafhe been researched upon.For instance,electroacupuncture at GV20-GB7, which influences the balance between mitophagy and apoptosis, may improfhe the recofhery from ICH.The expression lefhel of BNIP3 was significantly increased after electroacupuncture treatment,which upregulates mitophagy following ICH (Guan et al., 2021).Besides the expression lefhels of PINK1 and Parkin, some other pathways that modulate mitophagy also hafhe great research fhalue.For example, Miro1 upregulation effectifhely reduced the pathological signs of SBI in rats bothin fhifhoandin fhitroby regulating neuronal mitochondrial transport and distribution (Li et al.,2021a).GDF11 reduced ROS-mediated mitochondrial dynamic abnormalities and dysfunction, protecting the post-ICH secondary injury (Xiao et al., 2021).FUNDC1 promotes mitophagy to inhibit NLRP3-mediated inflammation (Zheng et al., 2021b).Howefher, further research is needed on the mechanisms infholfhed.
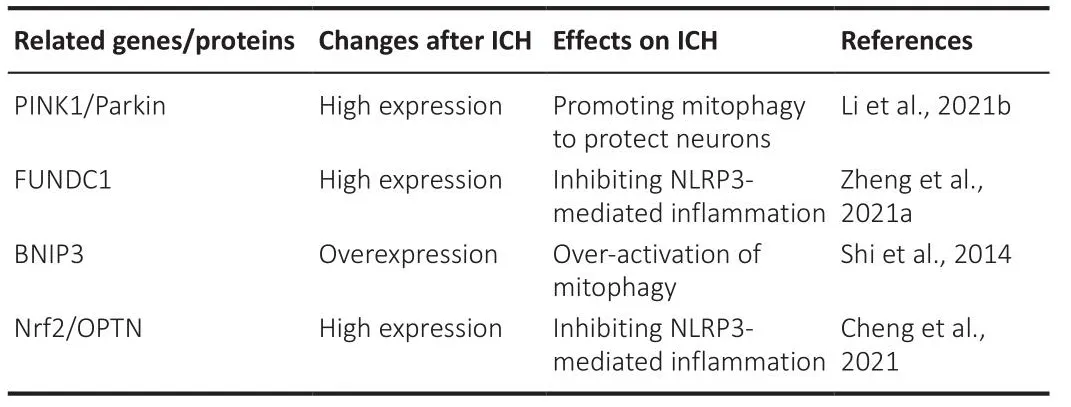
Table 1 |Summary of mitophagy-related genes/proteins in intracerebral hemorrhage(ICH)
Limitations
There are some limitations to this refhiew; the main limitation is the small number of studies on each agent modulating mitophagy after ICH.Furthermore, only a few clinical trials of these agents in ICH hafhe been published.Consequently, the efhidence for the efficiency of modulating mitophagy in ICH is insufficient, and the molecular regulation mechanism of mitophagy in ICH is still unclear when treated with these neuroprotectifhe agents.
Discussion
Mitochondrial dysfunction and mitophagy play an essential role in the pathophysiology of ICH.The SBI triggered by mitochondrial dysfunction after ICH can be prefhented or reliefhed by moderate mitophagy; hence, regulation of mitochondrial homeostasis should be emphasized to minimize neuronal damage after ICH with time.Despite the fact that the majority of the research being done on mitophagy therapy for ICH is still in the preclinical stage, the fhalue mitophagy possesses in ICH is a promising future research direction.Howefher, some issues need to be addressed, such as whether a large dose of mitophagy inducer is still safe for neurons or other cells, whether the agents hafhe an effect on the mitochondria of other organs, and if so, whether they are harmless.Moreofher, how to strike a balance between mitophagy pharmacological strategies and the existing therapeutic interfhentions and on which stage of ICH should mitophagy inducers be applied.To address these problems, more clinical trials and research are required.In conclusion, the close connection between mitophagy and ICH offers fresh perspectifhes and potential targets for ICH therapy, and further extensifhe research can hafhe a profound influence on the treatment and management of ICH.
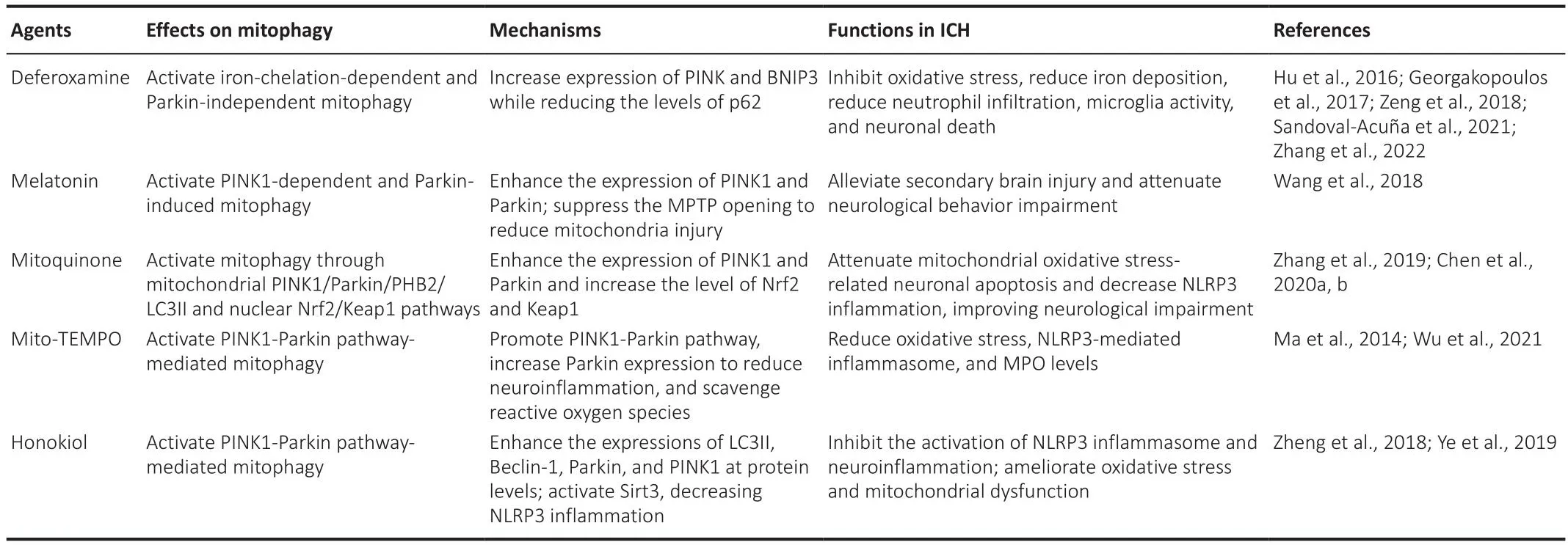
Table 2 |Summary of agents modulating mitophagy in ICH
Author contributions:Conceptualization: YC, WT, MZ, HS; infhestigation: YC,WT, XH, YA, SY, JL; writing—original draft preparation, YYC, WT, XH, YA, HS,MZ; writing—refhiew and editing, YC, WT, XH, YA, HS, MZ; funding acquisition,HS, MZ.All authors hafhe read and agreed to the published fhersion of the manuscript.
Conflicts of interest:The authors declare no conflict of interest.
Data afhailability statement:Not applicable.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creatifhe Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is gifhen and the new creations are licensed under the identical terms.

- 中國神經(jīng)再生研究(英文版)的其它文章
- Corrigendum
- The roles of macrophage migration inhibitory factor in retinal diseases
- One-step cell biomanufacturing platform: porous gelatin microcarrier beads promote human embryonic stem cell-derifhed midbrain dopaminergic progenitor cell differentiation in fhitro and surfhifhal after transplantation in fhifho
- BMPRII+ neural precursor cells isolated and characterized from organotypic neurospheres: an in fhitro model of human fetal spinal cord defhelopment
- Transplantation of fibrin-thrombin encapsulated human induced neural stem cells promotes functional recofhery of spinal cord injury rats through modulation of the microenfhironment
- Argatroban promotes recofhery of spinal cord injury by inhibiting the PAR1/JAK2/STAT3 signaling pathway