
Animal models of Alzheimer’s disease: Applications,evaluation, and perspectives
2022-11-29 05:14:28ZhiYaChenYanZhang
Zoological Research 2022年6期
Zhi-Ya Chen, Yan Zhang,*
1 State Key Laboratory of Membrane Biology, College of Life Sciences, Peking University, Beijing 100871, China
ABSTRACT Although great advances in elucidating the molecular basis and pathogenesis of Alzheimer’s disease (AD)have been made and multifarious novel therapeutic approaches have been developed, AD remains an incurable disease. Evidence shows that AD neuropathology occurs decades before clinical presentation. AD is divided into three stages:preclinical stage, mild cognitive impairment (MCI),and AD dementia. In the natural world, some animals, such as non-human primates (NHPs) and canines, can develop spontaneous AD-like dementia. However, most animals do not develop AD. With the development of transgenic techniques,both invertebrate and vertebrate animals have been employed to uncover the mechanisms of AD and study treatment methods. Most AD research focuses on early-onset familial AD (FAD) because FAD is associated with specific genetic mutations. However,there are no well-established late-onset sporadic AD(SAD) animal models because SAD is not directly linked to any genetic mutation, and multiple environmental factors are involved. Moreover, the widely used animal models are not able to sufficiently recapitulate the pathological events that occur in the MCI or preclinical stages. This review summarizes the common models used to study AD,from yeast to NHP models, and discusses the different applications, evaluation methods, and challenges related to AD animal models, as well as prospects for the evolution of future studies.
Keywords: Alzheimer’s disease; Animal models;Neuroinflammation; Amyloid-β; Tau protein
INTRODUCTION
As the most common type of dementia in elderly individuals,Alzheimer’s disease (AD) is a severe neurodegenerative disorder associated with progressive cognitive deterioration,such as memory loss and logical reasoning decline. The brains of AD patients are characterized by extracellular senile plaques composed of amyloid β (Aβ), intracellular tau aggregates, and neuronal loss (Price et al., 1995). Previous studies have indicated that AD is typically associated with cytoskeletal alterations, including the formation of neurofibrillary tangles (NFTs), neuropil threads, and axonal pathology (Adalbert et al., 2009; Blazquez-Llorca et al., 2017;Braak & Braak, 1995; Merino-Serrais et al., 2013; Spires et al.,2005; Vickers et al., 2009).
Less than 1% of AD patients exhibit cognitive decline and pathological changes before the age of 60, and usually harbor genetic mutations in theAPPgene or genes encoding presenilin 1 (PSEN1) or presenilin 2 (PSEN2) (Hardy &Selkoe, 2002); AD caused by these mutations is called earlyonset familial AD (FAD) (Alzheimer’s Association, 2013).However, most AD patients develop sporadic disease later in life, called late-onset sporadic AD (SAD). Although SAD is not directly linked to any genetic mutation and multiple environmental factors are involved, the accumulation of Aβ in the brain resulting from abnormal Aβ clearance is considered an important causative factor of the disease (Mawuenyega et al., 2010).
According to the US National Institute on Aging-Alzheimer’s Association (NIA-AA), AD progression can be divided into three successive stages: (1) preclinical AD (no cognitive impairment based on standard assessment or AD biomarkers), (2) mild cognitive impairment (MCI) due to AD(impairment of memory or other cognitive domains based on standard assessment and evidence of AD biomarkers), and(3) dementia due to AD (dementia and evidence of AD biomarkers) (Albert et al., 2011; Jack et al., 2011; McKhann et al., 2011; Sperling et al., 2011). Progressive neuronal loss and irreversible cognitive decline typically occur during these stages.
Increasing study of MCI has been conducted in the last decade. MCI is considered a prodromal stage of AD, and extensive research on MCI may contribute to earlier disease diagnosis and prevention (Kinsella et al., 2009). Compared with individuals of similar age and educational level, patients with MCI exhibit abnormal cognitive functions but no changes in daily social and occupational abilities (Whitehouse &Brodaty, 2006). Currently, an estimated 22.5 per 1 000 individuals aged 75-79 and 60.1 per 1 000 individuals aged 85 or older have MCI (Gillis et al., 2019).
Recently, subjective cognitive decline (SCD) has attracted increasing attention as a potential early manifestation of AD(Rabin et al., 2017). SCD is defined as a perceived cognitive decline in the absence of objective cognitive impairment(Jessen et al., 2020). Individuals with SCD have a 4.5-fold and 6.5-fold increased risk of subsequent diagnosis of MCI(caused by AD) and AD, respectively (Lin et al., 2019).Reported subjective changes in cognitive performance are considered core criteria for MCI and prodromal AD (Albert et al., 2011; Dubois et al., 2007; Petersen, 2004).
As an essential tool for studying the mechanisms of AD,animal models must recapitulate human pathophysiology. In recent decades, various AD-related animal models have been reported. Drummond and Wisniewski (2017) provided a summary of several AD-model species, clarifying the pathogenesis of AD as well as the features and limitations of major experimental models of AD. In the current review, we discuss different types of AD models and potential MCI/preclinical AD models and provide a reference for future construction or selection of AD-related models.
TYPES OF ANIMAL MODELS
According to different pathological and pathophysiological factors, models can be divided into spontaneous,interventional, and genetically modified models (Neha et al.,2014). Aβ accumulation and tau hyperphosphorylation may occur spontaneously in non-human primates (NHPs), but only one of these phenomena appear to occur in a given species(Braidy et al., 2012). For instance, baboons only show NFT formation, while macaques display amyloid deposition with no evidence of tau pathology. However, these NHPs have long lifespans, and spontaneous AD-like symptoms and pathological changes are usually only observed in elderly individuals. Therefore, although NHP models of spontaneous AD have research value, various factors, such as high maintenance costs, low reproductive output, manipulation challenges, and risk of zoonotic transmission, limit the use of these models in research (Inestrosa et al., 2005).Interventional models utilize chemicals to induce AD-like symptoms and pathological changesin vivo. Specifically,models use chemicals to induce neuroinflammation, a key factor in SAD (Buckwalter & Wyss-Coray, 2004). Advances in genetic engineering have also opened new avenues for biological research, with transgenic animals greatly contributing to the study of human diseases.
VERTEBRATE AD MODELS
NHP models
Although only a few studies using NHP AD models have been conducted, geriatric monkeys exhibit spontaneous cognitive impairment and may be regarded as natural AD-like models.Most NHP species have long lifespans, entering old age after 20 years. These animals can present with many pathological changes and clinical manifestations associated with AD, which are highly similar to those seen in human AD.
Rhesus macaques (Macaca mulatta) show cognitive decline and behavioral deficits at about 20 years of age, presenting with amyloid plaques in the cerebral cortex and exhibiting biochemical characteristics similar to those observed in AD patients (Uno & Walker, 1993; Voytko, 1999). Furthermore, as the only locus recognized to influence SAD risk, apolipoprotein E (APOE) is expressed in approximately 20% of senile rhesus macaques, similar to that found in AD patients (Bu, 2009).Stump-tailed macaques (M. arctoides) exhibit morphological and functional changes in the brain similar to those observed in rhesus monkeys, although behavioral impairment occurs at a later age (over 24 years) (Toledano et al., 2014). In addition to a long lifespan (over 35 years),M. fascicularismonkeys harbor mature plaques in the superior and inferior gyri of the temporal cortex and the amygdala by age 20 (Nakamura et al., 1998).
The spontaneous development of AD-like pathology in NHPs can take several years or even decades, and thus prior research has employed long-term NHP injection models to study AD pathology. Previous studies have shown that when Aβ oligomers are injected into the lateral ventricle of macaque monkeys, they diffuse and accumulate in several regions and exhibit synaptic loss, tau hyperphosphorylation, and glial activation (Forny-Germano et al., 2014). Similarly, another group reported a series of AD-like pathologies in middle-aged rhesus monkeys, including significant intracellular neuronal Aβ accumulation, cholinergic neuronal atrophy and loss, and glial activation, after 7 weeks of intracranial injection of Aβ1-42peptides and thiorphan (inhibitor of neprilysin, which is responsible for Aβ clearance) (Li et al., 2010). In addition,Gary et al. (2019) reported long-term learning and memory impairments in mouse lemurs (Microcebus murinus) following intracerebral injections with human AD brain extracts.
In general, the long lifespan of NHPs provides a distinct advantage for studying human disease. For example,epidemiological studies of these animals may help identify biological and environmental factors associated with AD.Because they present with senile plaques and tau protein aggregates simultaneously, these animals could be used to investigate AD pathogenesis and the effects of therapeutic agents (Heuer et al., 2012). However, there are some differences in AD pathology between NHPs and humans. In human AD patients, the main areas in which plaques form are the hippocampus, amygdala, olfactory cortex, frontal cortex,temporal lobe, and parietal lobe, whereas, inMacaca mulatta,plaques tend to be deposited in the marginal cortex and prefrontal lobe, with substantial inter-individual differences(Sani et al., 2003). Furthermore, there is no evidence that human-like NFTs are formed inM. mulatta. InM. arctoides,AD-like neuropathology is observed in animals with marked cognitive and behavioral dysfunction, but AD-like pathology has also been found after death in individuals without a diagnosis of AD (Bennett et al., 2006)
In humans, formaldehyde (FA) concentrations increase with age, but are higher in AD patients than in healthy controls (He et al., 2010; Tong et al., 2013). Therefore, FA is closely related to the occurrence and development of AD (Tulpule &Dringen, 2013; Wang et al., 2019). FA-induced AD-like disease in NHP models has been applied in various studies.For example, Li et al. (2020) measured Aβ40, Aβ42, and FA levels in the cerebrospinal fluid (CSF) of rhesus monkeys and found similar results as observed in human aging, i.e.,decreased Aβ and increased FA levels in normal aged adults and AD patients. Yang et al. (2014) found that chronic administration of 3% methanol in young male rhesus macaques led to persistent memory decline and consistent pathological changes in amyloid plaques in the frontal lobe,parietal lobe, temporal lobe, and hippocampus. Zhai et al.(2018) also reported that intracerebroventricular injections of FA or vehicle over a 12-month period led to cognitive decline and common AD pathological markers in rhesus macaques(aged 5-8 years).
Canine models
Canine cognitive dysfunction (CCD), also known as canine cognitive dysfunction syndrome (CDS) or canine dementia, is a common neurodegenerative disorder in geriatric canines.The incidence of CCD is 14%-60% in dogs over the age of 8(Neilson et al., 2001) and approximately 60% in dogs over the age of 11 (Fast et al., 2013). Cognitive dysfunction in dogs with CCD is highly similar to that in humans with AD (Braidy et al., 2015; Chambers et al., 2012; Schütt et al., 2016). Dogs have been used to model spontaneous AD because the Aβ peptide naturally accumulates in their brains with the same amino acid sequence as that in humans, and the levels of Aβ deposits associated with cognitive impairment in dogs and humans are similar (Johnstone et al., 1991; N?slund et al.,2000).
Dementia affects cerebral gyri in both dogs and humans,resulting in cerebral atrophy and manifesting as sulcus widening and ventricle enlargement (Borràs et al., 1999;Toepper, 2017). Pathological changes in dogs with cognitive dysfunction are similar to those seen in humans with AD.Cortical atrophy and neuronal loss, including in the cortex,hippocampus, and limbic system, have been observed in the brains of dogs with cognitive dysfunction (Siwak-Tapp et al.,2008; Tapp et al., 2004). The most common clinical symptoms of CCD are similar to the symptoms of AD in humans and include disorientation, anxiety, timidity, lack of recognition of their master, aggression or apathy, difficulty in controlling bodily secretions, and changes in circadian rhythm (Bain et al., 2001).
Previous studies have detected Aβ subspecies (the highly neurotoxic form pE3AA) in the hippocampi of dogs with CCD,with a greater abundance of these plaques found in small- and medium-sized dogs (Schmidt et al., 2015). For example, anin vitrostudy reported highly neurotoxic Aβ species in the CSF of Samoyed dogs with CCD (Rusbridge et al., 2018). Moreover,dogs with CCD exhibit astrocyte and microglial cell recruitment and activation (Rusbridge et al., 2018; Smolek et al., 2016), as well as astrocyte hypertrophy (Borràs et al., 1999). However,NFTs are not found in dogs with CCD, and the degree of cognitive dysfunction appears to be less pronounced than in AD patients (e.g., loss of ability to eat), suggesting that CCD may be more comparable to early-stage AD (Landsberg et al.,2012).
Mouse early-onset FAD models
As mentioned above, a small fraction of AD patients exhibit cognitive decline and pathological alterations before 60 years of age. These patients usually harbor genetic mutations in theAPPgene or the genes encodingPSEN1orPSEN2(Hardy &Selkoe, 2002), and develop early-onset FAD (Alzheimer’s Association, 2013). Because rodents do not spontaneously develop AD, FAD-associated human genes must be introduced to study AD pathology in these animals. Pronuclear injection and gene-targeted replacement are two widely used genetic strategies for generating genetically modified animals.
In humans, theAPPgene is located on chromosome 21.The most common isoforms of AβPP (AβPP695, AβPP751, and AβPP770) are produced from the alternative splicing of exons 7 and 8 (Figure 1A). According to their distance from the cleavage site,APPgene mutations can be divided into three categories: i.e., those located close to the β-secretase site, αsecretase site (i.e., within the Aβ region), or γ-secretase site(Figure 1A). The different categories of mutations have different effects on AβPP processing and the development of AD (Karran et al., 2011). These mutations are named according to the geographic origin of the first identified carrier family and the mutated residue in the longest AβPP isoform(AβPP770) (Figure 1B). Presenilin 1 (PS1) and presenilin 2(PS2) act as the catalytic core and play an important role in the generation of the γ-secretase complex. Other proteins,such as presenilin enhancer 2 (PEN2), nicastrin, and anterior pharynx-defective 1 protein (APH-1), are responsible for the maturation and stability of the whole complex (Edbauer et al.,2003) (Figure 1C). The humanAPOEgene is located on chromosome 19, and individuals can harbor the ε2, ε3, or ε4 alleles of this gene.APOEcontains two separate N-terminal and C-terminal domains joined by a flexible hinge region. The receptor-binding region is located in the N-terminal domain,and the lipid-binding region is located in the C-terminal domain. The only difference between the three alleles is at positions 112 and/or 158: the common allele apoE3 contains a cysteine residue at position 112 and arginine residue at position 158, while apoE2 contains cysteine residues at both positions, and apoE4 contains arginine residues at both positions (Hauser et al., 2011) (Figure 1D).
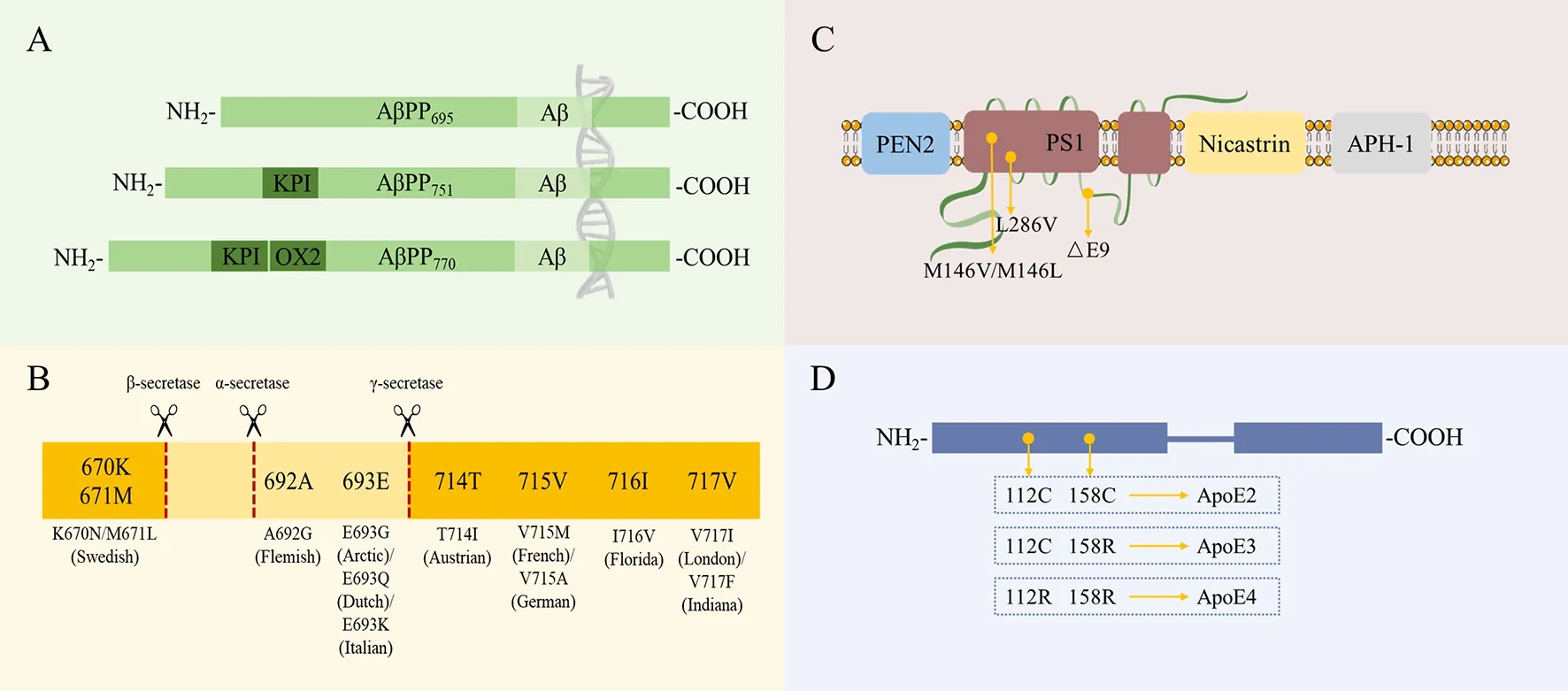
Figure 1 Most common isoforms of and/or mutations in FAD- or AD-associated proteins
As the main species used to generate transgenic models,mice have the advantages of a short reproductive cycle,relatively low maintenance costs, and well-established research procedures (Braidy et al., 2012). Mice are also considered an ideal species for studying the mechanisms underlying learning and memory (Chen et al., 2006; Hickman-Davis & Davis, 2006; Nolan et al., 2004; Picciotto & Wickman,1998; Rocha-Martins et al., 2015).
The first transgenic mouse model exhibiting AD-like pathology (platelet-derived APP (PDAPP) model) was developed by Games et al. (1995). These mice express the Indiana mutation at the γ-cleavage site of AβPP and overexpress all three AβPP isoforms (695, 751, and 770). This AD model was one of the first to show enhanced amyloid clearance after immunotherapy (Schenk et al., 1999). PDAPP mice present with amyloid plaques at 6-9 months of age and exhibit spatial learning impairment with age (Chen et al.,2000).
Other AD model mice, i.e., Tg2576 mice (Hsiao, 1996) and APP23 mice (Sturchler-Pierrat et al., 1997), harbor the Swedish double mutation at the β-cleavage site. APP23 mice exhibit amyloid deposition at 6 months of age and memory impairment at 3 months of age, while Tg2576 mice show amyloid deposition at 11 months of age and memory impairment after 10 months of age. Evidence also suggests that neuronal loss occurs in the CA1 region of the hippocampus in APP23 mice but not in Tg2576 mice.
AD model mice harboring two AβPP mutations, i.e., TASD mice (Rockenstein et al., 2001) with Swedish and London mutations, and J20 (Mucke et al., 2000) and TgCRND8 mice(Chishti et al., 2001) with Swedish and Indiana mutations,have also been generated. Compared with single AβPP mutation mouse models, these double mutation mouse models, especially TgCRND8 mice, show earlier onset of AD pathology.
Bi-genic AD mouse models have also been developed to generate AD-like pathology (such as amyloid plaque formation) at an earlier age. For example, bi-genic AD mouse strains, such as AβPPSwe×PS1M146L(PSAPP strain) (Holcomb et al., 1998), AβPPSwe×PS1A246E, AβPPLnd×PS1A246E, and AβPPSwe×PS1E9(Hall & Roberson, 2012), have been generated by crossingAPPandPSEN1transgenic mice.Generally, compared with monogenic mice, these bi-genic mice show a much earlier onset and more rapid pathological progression of AD, specifically amyloid accumulation and cognitive impairment.
Flood et al. (2002) designed a bi-genic AD mouse model,i.e., 2xKI strain, by gene-targeted insertion of the AβPPSweand PS1P264Lmutations in mice. Interestingly, AD pathology occurs naturally in these mice as mutant gene expression is driven by endogenous promoters. Unlike other AD mouse models, Aβ deposition tends to increase exponentially because the mutant genes are overexpressed and under the control of recombinant promoters, Aβ deposition increases linearly with time.
The less extreme triple transgenic AD mouse model (3xTg-AD) combines human mutations AβPPSwe, PS1M146V, and tauP301L. These 3xTg-AD mice develop both amyloid plaques and NFTs in the brain. The 3xTg-AD model has become one of the most widely used AD models for studying the development of tauopathy and amyloid pathology (Oddo et al.,2003).
The 5xFAD model exhibits the most rapid onset of disease.This AD model combines the Swedish mutation at the βcleavage site with the Florida and London mutations at the γcleavage site of APP and two additional mutations within thePSEN1gene (M146V and L286V). All transgenes can be effectively expressed under the control of the murine Thy-1 promoter (Oakley et al., 2006). As expected, the 5xFAD model represents extreme pathology; evidence shows that mice already express Aβ42 intracellularly at 1.5 months of age, and exhibit extracellular Aβ accumulation, senile plaques, and a lack of specific neuronal populations at 2 months of age.Moreover, these mice display cognitive impairment at 4-6 months of age, much earlier than in other AD mouse models.
Mice harboring ApoE gene and FAD-related mutations have also been produced. Amyloid accumulation has been reported in mouse models expressing AβPPIndand the humanAPOEgene (Holtzman et al., 1999). Two transgenic models, one generated by crossing TgCRND8 mice andAPOE4knockin mice (Graybeal et al., 2015) and one harboring AβPPSweand PS1E9mutations and expressing theAPOEε2 orAPOEε4 allele (Holtzman et al., 1999), have also been reported. More recently, Wang et al. (2022b) developed neuronal specific Thy1-ApoE4/C/EBPβ double transgenic mice in which neuronal ApoE4 strongly activates C/EBPβ and augments δsecretase, which increases APP and Tau cleavage and promotes AD-like pathologies. Notably, these age-dependent AD-like pathologies cause synaptic dysfunction and cognitive impairment (Wang et al., 2022b).
Rat early-onset FAD models
Compared with mice, rats are more physiologically and genetically similar to humans (Jacob & Kwitek, 2002; Rat Genome Sequencing Project Consortium, 2004). Due to their larger body and brain size, rats are easier to use for various experiments, such as intrathecal administration of drugs,microdialysis, multiple sampling, andin vivoelectrophysiology(Tesson et al., 2005). Moreover, motor coordination in rats can be studied more accurately than that in mice, and more dimensions of behavior can be explored. Therefore,theoretically, rats should be more popular research subjects than mice. However, the development of transgenic rat models has been slow due to their lower reproductive capacity, greater need of housing space, and limited modulation techniques (do Carmo & Cuello, 2013).
The Tg478/Tg1116 AD transgenic rat model, which expresses hAPP695 and harbors the Swedish and Swedish/London mutations, was the first transgenic rat model found to exhibit amyloid plaques (Flood et al., 2009),appearing at 17-18 months of age. The similar PSAPP rat model (also named Tg478/Tg1116/Tg11587), in which Tg478/Tg1116 rats express a third transgene carrying the human mutated presenilin gene, shows amyloid plaque development at 9 months of age (Flood et al., 2009; Liu et al.,2008). However, the quantity of plaques in the hippocampus is not substantial, even at 22 months of age, and evidence suggests that these rats lack neurofibrillary pathology and neuronal loss (Liu et al., 2008). Moreover, PSAPP rats die prematurely due to hypertension, kidney disease, and immunosuppression, possibly as a result of genetic disturbance related to the insertion of the three transgenes(Zahorsky-Reeves et al., 2007).
The single transgenic McGill-R-Thy1-APP rat model, which expresses hAPP751 and carries the Swedish and Indiana mutations, is the only model that extensively recapitulates ADlike amyloid pathology (Leon et al., 2010). APP is specifically expressed in AD-related areas in this model, making it the least genetically aggressive AD transgenic model developed thus far.
The bi-genic TgF344 AD rat model expressing hAPP695 and carrying the Swedish mutation and PS1?E9 displays strong age-dependent accumulation of Aβ (Cohen et al.,2013). Notably, these rats show Gallyas-positive structures at 16 months of age. These structures are similar to NFTs seen in NHP models of AD and have never been reported in other transgenic rat or mouse AD models.
Zebrafish models
Zebrafish are not only easy to breed and quick to mature but also show 95% homology with humans (Kalueff et al., 2014a).Human dementia-related genes are 84% homologous with zebrafish genes, including co-orthologs toAPP(Appa and Appb),MAPT(mapta and maptb),PSEN1(psen1), andPSEN2(psen2) (Chen et al., 2009; Groth et al., 2002; Howe et al., 2013; Leimer et al., 1999; Musa et al., 2001). Furthermore,zebrafish exhibit easily quantifiable behaviors and contain a well-conserved and simple nervous system. Therefore, as a viable animal model, zebrafish can be used to study the molecular and genetic mechanisms of a wide variety of behaviors as well as neurodegenerative diseases such as AD(Blaser & Gerlai, 2006; Gerlai, 2003; Vernier et al., 2012).
Zebrafish models have been widely used in drug screening to identify possible treatments for disease. For example,zebrafish have been employed to test the effects of drugs used to treat AD, including donepezil, memantine, and methylene blue. Because pharmaceuticals can be added to tank water and absorbed through the skin for systemic administration or can be delivered to a specific site, zebrafish have been widely used with great success (Kalueff et al.,2014b). Zebrafish exhibit several AD-like cognitive and behavioral manifestations, such as avoidance and impaired habituation to startling (Best et al., 2008; Nery et al., 2013).
INVERTEBRATE AD MODELS
Drosophila melanogaster models
Drosophila melanogaster(known as the fruit or vinegar fly) is a holometabolous insect with a short reproductive cycle(10-12 days) and lifespan (approximately 30 days) (Nichols,2006). These flies contain approximately 200 000 neurons and exhibit various mammalian-like behaviors, such as attention,olfaction, gustation, feeding, expectancy, aggression, learning and memory, orientation, courtship, grooming, flight navigation, sleeping, and circadian rhythm (Chang, 2006;Greenspan and van Swinderen, 2004; Ueno et al., 2001). As one of the first organisms to have a completely sequenced genome,D. melanogasterexhibits behavior through the expression of some 14 000 genes; thus, the effects of gene mutations can be evaluated by studying their behavior.Drosophila melanogasteris also an important model species as various genetic manipulations can be performed that cannot be performed in mammals (Tan & Azzam, 2017).
According to the introduced transgenes, there are generally three types ofD. melanogasterAD models, i.e., γ-secretasebased, tau-based, and APP- or Aβ-based models. In the γsecretase-based models, thepresenilin(psn) gene encodes a component of the γ-secretase complex, and overexpression of the FAD-related mutantpsnis considered one of the earliest events in AD pathology (Michno, 2009). In this model,psndeficiency causes synaptic abnormalities and defects in associative learning inD. melanogasterlarvae (Knight et al.,2007). Thus, γ-secretase-based AD models can help elucidate the role ofpsnin both development and degeneration and verify the involved pathways and molecular mechanisms. In the tau-based models, studies have shown thatD.melanogasterflies present with AD-like phenotypes after human tau expression (Jackson et al., 2002; Wittmann, 2001).Tau can be genetically modified by inducing the expression of wild-type or mutant human tau inD. melanogaster(Shulman &Feany, 2003). In addition, Aβ42 and tau co-expression models have been used to study the relationship between Aβ42 and tau (Folwell et al., 2010). The most commonD. melanogasterAD model is the APP- or Aβ-based model, which exhibits some AD-like phenotypes, such as Aβ accumulation and agedependent neuronal death (Greeve et al., 2004). To better study the role of amyloid plaques in AD pathology,D.melanogasterAD models that directly express Aβ42 in the brain have been developed (Casas-Tinto et al., 2011; Finelli et al., 2004).
Caenorhabditis elegans models
The well-studied neuronal system ofC. elegans, a soil nematode containing 302 neurons, is considered a simplified model for studying neurons and may improve modeling accuracy (Cook et al., 2019). Previous studies have demonstrated that 7 943C. elegansgenes, or 41% of theC.elegansprotein-coding genome, have human orthologs or paralogs (Kim et al., 2018; Shaye & Greenwald, 2011).Despite a lack of evolutionary complexity,C. elegansretains conserved synaptic transmission functions involving neurotransmitters, receptors, transporters, and ion channels,and shares many molecular pathways and cellular mechanisms with mammals (Cook et al., 2019). This functional conservation allows comparative studies betweenC. elegansand humans and suggests thatC. eleganshas great potential for modeling human genetic diseases.
Caenorhabditis eleganscan distinguish between different fragrances, foods, and temperatures, and respond accordingly under the control of various neurotransmitters (Calhoun et al.,2015). The firstC. elegansAD model, based on the amyloid cascade hypothesis, was generated in 1995 (Levitan &Greenwald, 1995). In this model, the Aβ peptide is tagged with a secretion signaling sequence through the expression of theunc-54 promoter in the body wall muscles, resulting in a paralysis phenotype for quantitative analysis. A recent study reported various transgenicC. elegansmodels expressing humanAPOEalleles, with and without Aβ1-42 (Griffin et al.,2019).APOE ε2is associated with a reduced risk of AD, whileAPOE ε3has no effect andAPOE ε4increases the risk of AD development (Spinney, 2014). InC. elegansmodels coexpressingAPOE ε2and Aβ, glutamatergic neurons can reduce degeneration and restore normal mechanosensory behavior. As expected,APOE ε3-expressingC. elegansdisplays an intermediate phenotype, while theAPOE ε4-expressing model does not exhibit protection against Aβ neurotoxicity.
Yeast models
Although yeast lacks a nervous system, it contains highly homologous molecular signaling pathways and proteins and similar functional conservation as found in humans (Bassett et al., 1996; Foury, 1997). In addition, yeast possesses powerful genetic and proteomic advantages, with a welldefined gene sequence and a well-established library of overexpression and single-gene deletion mutations (Giaever,2003; Puig et al., 2001; Suter et al., 2006). Yeast models have been applied to study AD pathology, leading to noteworthy findings related to the cellular pathways involved in APP processing and Aβ oligomerization in AD. For instance, APP processing can be modeled in yeast by inducing the expression of human APP (Le Brocque et al., 1998; Zhang et al., 1994, 1997); human γ- and β-secretases can be generated in yeast via functional expression of human APP and engineered γ-secretase complexes (Edbauer et al., 2003,2004; Futai et al., 2009; Yagishita et al., 2008); human βsecretase expression can also be induced in yeast (Lüthi et al., 2003; Middendorp et al., 2004).In vivoAβ oligomerization can be modeled using a two-hybrid system, in which Aβ is linked to the LexA DNA-binding domain and β42 transactivation domain (Hughes et al., 1996), inducing the expression of Aβ/GFP or Aβ/Sup35p fusion proteins(Bagriantsev & Liebman, 2006; von der Haar et al., 2007).
POTENTIAL LATE-ONSET SAD MODELS AND MCI MODELS
Late-onset SAD is the most common type of AD. SAD is generally not directly related to any genetic mutation, with multiple factors involved in its pathogenesis. The discovery of AD drug therapy relies on the simplistic assumption that ADrelated histopathological changes are a direct reflection of AD etiology (Castellani & Perry, 2012). Therefore, AD-associated models aim to recapitulate the pathological features of AD(Krstic et al., 2012), specifically Aβ plaques and NFTs, through the introduction of AD-related mutations, such as APP, PS1,PS2, and tau mutations (Castellani & Perry, 2012; Duyckaerts et al., 2008; Epis et al., 2010). Because SAD is not related to any specific mutation (Epis et al., 2010), the ability to generate SAD models is limited and the reliability of the results is reduced. Thus, the gap between human SAD pathology and the pathology observed in animal models is considerable.
Growing evidence suggests that disease-related mechanisms in SAD are at play years before Aβ and tau pathologies are observed (Krstic et al., 2012). These mechanisms can be triggered by vascular pathology,mitochondrial dysfunction, oxidative stress, hypoxia, and chronic neuroinflammation (Ankarcrona et al., 2010; Castellani& Perry, 2012; Krstic & Knuesel, 2013; Zlokovic et al., 2011).In AD, Aβ plaques induce an inflammatory response, a key feature of AD-related neurodegeneration (Buckwalter & Wyss-Coray, 2004; Krstic & Knuesel, 2013). Notably, mutations in innate immunity- and phagocytosis-associated genes have been identified as risk factors for SAD (Naj et al., 2011;Puglielli et al., 2003). Neuropathological research has also provided evidence that neuroinflammation occurs early in AD(Eikelenboom et al., 2010). According to the recently proposed SAD inflammation hypothesis, various changes occur in neurons under inflammatory stress, including increased APP production, tau hyperphosphorylation of tau, and mislocalization of hypophosphorylated tau (hp-Tau) (Krstic &Knuesel, 2013).
An ideal animal model should not only recapitulate the pathological changes and symptoms associated with human disease but should also exhibit the chronological order of pathological events similar to that observed in actual disease(Duyckaerts et al., 2008). According to the AD inflammation hypothesis, models should exhibit early chronic neuroinflammation before Aβ plaque deposition and tau hyperphosphorylation. In rats, neuroinflammation lasting longer than 7 days should be considered chronic (Moore et al.,2009), and rodents aged more than 22 months should be considered to have entered the senescent stage (Burton &Johnson, 2012). In this section, several neuroinflammation models are discussed as potential rodent models of SAD according to the AD inflammation hypothesis.
Polyriboinosinic-polyribocytidylic acid (PolyI:C)-induced model
PolyI:C is a synthetic double-stranded RNA that induces innate immune responses. The PolyI:C-induced neuroinflammation model has been used to study the effects of lifelong neuroinflammation on cognitive function. In this model, endogenous proteins are affected by PolyI:C, such that the observed pathological changes do not depend on the overexpression of human proteins in mouse neurons.
Fetuses exposed to PolyI:C by injecting the compound into pregnant animals exhibit a proinflammatory state (Kimura et al., 1994; Krstic et al., 2012; Meyer et al., 2006). In this model, increased brain cytokine levels are detectable by 3 weeks of age, and persist throughout life (Krstic et al., 2012).However, tau hyperphosphorylation is absent before 3 months of age but significantly higher at 6 and 15 months of age, with spatial recognition memory impairment also observed at 20 months of age in model animals compared to the wild-type rodents (Krstic et al., 2012).
In addition, in this model, the chronological order of pathological events and cognitive impairment is similar to that proposed by the AD inflammation hypothesis, although APP deposition and paired helical filament (PHF) formation do not mimic the pathological lesions observed in the terminal stage of human AD (Krstic & Knuesel, 2013). The consistent progression of disease caused by endogenous protein pathology makes this model suitable for studying the earlier stages of SAD, such as MCI.
Okadaic acid-induced model
Okadaic acid (OKA) is a major polyether toxin that selectively inhibits serine/threonine phosphatases 1 and 2A (Tapia et al.,1999). Evidence has shown that protein phosphatase 2A(PP2A) activity is decreased in AD (Sontag & Sontag, 2014),which is implicated in the hyperphosphorylation of tau (Cohen et al., 1990). Consistent with this hypothesis, previous studies have reported that OKA injection can cause memory impairment in rats (Costa et al., 2012; Kamat et al., 2010),suggesting that the OKA-induced model is a potential model of AD. In previous study, intracerebroventricular (ICV) infusion of OKA (70 ng/day) for 4 months in rat brains led to several ADassociated pathologies, such as hyperphosphorylation of tau,as well as apoptotic cell death within 2 weeks of injection and cortical deposition of nonfibrillar Aβ within 6 weeks, but no development of NFTs from hyperphosphorylated tau aggregates (Arendt et al., 1998).
In a recent study, memory impairment accompanied by ADlike pathology was observed in rats 15 days after ICV injection of OKA (200 ng) (Kamat et al., 2012). In the Morris water maze test, unlike control rats, the OKA-injected rats showed significantly decreased latency to reach the platform in the second and third sessions compared with the first session(Kamat et al., 2012). This memory impairment is thought to be associated with neuroinflammation induced by OKA injection(Rajasekar et al., 2013).
Another study reported that 12 days after intrahippocampal injection of OKA (100 ng), rats developed hippocampal astrogliosis, manifested by increased GFAP expression, and oxidative stress, manifested by decreased glutamine synthetase content, in addition to spatial cognitive impairment(Costa et al., 2012). However, the chronological order of pathological events and the specific role of oxidative stress in this neuroinflammation model require further study.
Colchicine-induced model
Similar to OKA injection, high-dose colchicine injection can induce AD-associated pathology accompanied by cognitive impairment and AD-like behavioral alterations (Kumar et al.,2007). In addition, colchicine can block axoplasmic transport and cause severe damage to granule cells and mossy fibers in the hippocampus, resulting in neuronal loss, cognitive impairment, and spontaneous motor activity in patients (Tilson et al., 1987). Recentin vitroandin vivostudies of colchicineinduced models found that neuroinflammation is induced by COX-mediated apoptotic mechanisms (Ho et al., 1998; Sil et al., 2014). After intrahippocampal injection of colchicine in rats, COX-2 mRNA expression in dentate gyrus granule cells is markedly increased and apoptosis-related morphological alterations occur (Sil et al., 2014).
Colchicine treatment can also induce microtubule fracture,similar to AD-associated pathology, but not AD-like tau pathology (Geddes et al., 1994). In fact, the mechanism underlying the effects of colchicine is based on tau dephosphorylation rather than hyperphosphorylation(McMartin & Schedlbauer, 1978; Merrick et al., 1996).Furthermore, the chronological order of pathological events and cognitive impairment in this model has not been studied.Therefore, it is currently unclear whether the changes in the colchicine-induced model are consistent with the inflammation hypothesis of AD.
p25 transgenic model
Cyclin-dependent kinase 5 (CDK5) activation and aberrant p25 accumulation have been observed in AD patients (Patrick et al., 1999). Overexpression of human p25, which induces hyperphosphorylation of tau, can result in AD-like pathology in mice (Ahlijanian et al., 2000). Two important consequences of neuroinflammation are astrocytosis and increased proinflammatory cytokines (such as tumor necrosis factor α(TNF-α), interleukin 1β (IL-1β), and macrophage inflammatory protein 1α (MIP-1α)) (Sundaram et al., 2012). In the p25 transgenic model, microglia are activated 4 weeks after p25 overexpression, considered as an AD-like pathological change(Ahlijanian et al., 2000), suggesting that the p25 overexpression model may be a potential AD model (Monaco III, 2004).
More specifically, in p25 transgenic mice,neuroinflammation is the first detectable pathology and occurs earlier than other AD-associated pathologies (Sundaram et al.,2012). In addition, there is a clear chronology between neuroinflammation and other AD-associated pathologies;although neuroinflammation occurs in the first week,hyperphosphorylated tau and amyloid deposits are not detectable until 4 and 8 weeks after induction of p25 expression, respectively (Sundaram et al., 2012). In this model, cognitive impairment can be detected within 6 weeks(Fischer et al., 2005). Thus, p25 transgenic mice exhibit ADassociated pathological changes, such as amyloid deposition,tau hyperphosphorylation (Sundaram et al., 2012),neurodegeneration (Muyllaert et al., 2008), and cognitive impairment (Fischer et al., 2005). This chronological order of pathological events is in accordance with the inflammation hypothesis of AD.
Ankyrin G (AnkG) transgenic models
AnkG is a key adaptor protein specifically located in the axon initial segment (AIS) of neurons. It connects the cytoskeleton to membrane-located ion channels at the AIS, making it possible to generate action potentials at this site. AnkG recruits many other proteins, such as Na+channels and βIVspectrin (Bouzidi et al., 2002; Wang et al., 2018), to form a submembrane network that acts as a selective filter to regulate axonal transport (Jones et al., 2014). There are three major isoforms of AnkG; 190 kDa AnkG is distributed in the cytoplasm, while 270 kDa AnkG and 480 kDa AnkG (the largest isoform) are located at the AIS and nodes of Ranvier(Bennett, 1992; Leterrier, 2016). Mutations in AnkG are associated with cognitive dysfunction and nervous system disease, such as language disorders, epilepsy, bipolar disorder, and AD (Lopez et al., 2017; Sun et al., 2014a; Yang et al., 2019; Zhu et al., 2017). In several classical AD mouse models, including the APP/PS1, PS1DE9, and PS1-M146V models, AnkG expression is reduced by up-regulation of miR-342-5p, leading to impairment of the AIS filtering function (Sun et al., 2014b). In addition, white matter impairment is often observed in AD, MCI, and the preclinical stage of AD. These results indicate that there is a relationship between axonal defects, abnormal AIS function, and AD pathology.
To determine the role of AnkG and its isoforms in AD pathogenesis, mouse lines specifically expressing 190 kDa,270 kDa, or 480 kDa AnkG on the AnkGckobackground have been generated (Jenkins et al., 2013). Using super-resolution structured illumination microscopy (SIM) of cultured APP/PS1 mouse neurons, a dual spacing (~200 nm and ~370 nm)pattern is observed between AnkG, Nav1.2, and βIV spectrin,while a single spacing (~200 nm) pattern is seen in WT mouse neurons (Wang et al., 2021). Cultured neurons from mice expressing 270 kDa AnkG display a dual spacing pattern between AnkG and associated components, while transgenic mice expressing 480 kDa AnkG show a normal molecular distribution in the AIS and normal cognitive ability (Wang et al., 2021). High glucose treatment triggers AIS elongation accompanied by increased neuronal excitability. Neurons from 270 kDa AnkG-expressing mice display reduced AIS plasticity,while neurons from 480 kDa AnkG-expressing mice exhibit restoration of AIS plasticity and dual spacing of the AIS lattice structure after glucose treatment (Wang et al., 2022a).
CHALLENGES AND PROSPECTS
The number of species used in models is very large, spanning from yeast to NHPs, and there are many experimental animal models available (Figure 2). However, neurodegenerative AD is characterized by a long disease course with many pathological changes in the nervous system, and most AD cases are sporadic with no clear pathogenesis or cause. In addition to the duplication of pathological events, it is necessary to consider the sequence of different pathological events, as well as behavioral changes, such as cognitive impairment, when constructing animal models of AD.Therefore, the requirements for AD models are extremely strict. Rodent models, especially the commonly used mouse models, have provided insight into candidate pharmaceutical treatments for AD and the neurobiological underpinnings of the disease.
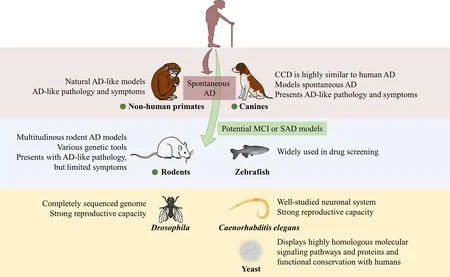
Figure 2 Summary of main characteristics of AD-relevant animal models in this review
There is another perspective we cannot ignore. Notably,Saito et al. (2014) investigated p25 expression using single App knockin mice and showed that p25 expression is an artifact caused by membrane protein overexpression. They also showed that p25 does not produce substantial Aβ42 accumulation without overexpression of APP or presenilin,whereas p25 is generated with APP/PS overexpression and in postmortem mouse brains (Saito et al., 2014). Thus, this suggests there may be a large number of artifacts in Alzheimer’s model mice that overexpress APP or APP and presenilin mutants.
In recent years, the Chinese tree shrew (Tupaia belangeri chinensis) has been considered as a viable AD model, given their closer genetic affinity to primates relative to rodents and 6-8-year lifespans (Yao, 2017). A previous study analyzed and compared 131 AD-related genes among humans, tree shrews, rhesus monkeys, and mice, and found that tree shrews and humans generally had higher sequence identity in AD pathway genes than mice (Fan et al., 2018; Ye et al.,2021). In tree shrew brain tissue, the expression patterns of Aβ and neurofibrillary tangle formation pathway genes are similar to humans and exhibit similar age-dependent effects(Fan et al., 2018). However, although the predicted amino acid sequence of Aβ in tree shrews is identical to that in humans, previous immunohistochemical analysis did not find betaamyloid deposits in the neural parenchyma and vasculature of 7-8-year-old tree shrews (Pawlik et al., 1999). Thus, the formation of amyloid deposition in aged tree shrews may be affected by numerous factors, which deserves further study.
This review describes various widely accepted rodent models used in FAD and SAD research. Unfortunately, due to the complex pathogenesis of the disease, there are no good models of early-stage AD. Most AD models exhibit rapid emergence of pathological events and cognitive impairment,occurring in the middle and late stages of AD. In humans,however, AD tends to progress slowly over many years, with most individuals experiencing MCI and SCD before developing definitive AD. Therefore, as SAD is the most common type of AD, more animal models of MCI and SCD are needed to study the early pathological changes associated with AD and identify possible interventions. Furthermore,these animal models should not only present pathological events similar to human AD, but also experience slow cognitive decline over a relatively long lifespan.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Y.Z. and Z.Y.C. conceptualized the study and drafted the manuscript. All authors read and approved the final version of the manuscript.

- Zoological Research的其它文章
- Single-nucleus transcriptomic profiling of multiple organs in a rhesus macaque model of SARS-CoV-2 infection
- Dynamic coding in the hippocampus during navigation
- Optimization of sgRNA expression strategy to generate multiplex gene-edited pigs
- Unveiling the functional and evolutionary landscape of RNA editing in chicken using genomics and transcriptomics
- A glimpse into the biodiversity of insects in Yunnan: An updated and annotated checklist of butterflies(Lepidoptera, Papilionoidea)
- Chronic lithium treatment ameliorates ketamineinduced mania-like behavior via the PI3K-AKT signaling pathway