
NoveI mutations in the BEST1 gene cause distinct retinopathies in two Chinese famiIies
2022-02-23 13:01:10ZhiHongZhuXinJinYiXinZhangRuiWangTongWuWeiLiuZeHuaChenHaiNanXieLanLanChenZiHaoLiuHouBinHuang
INTRODUCTION
Human bestrophin-1 (MIM 607854) is predicted to be a four-transmembrane protein consisting of 585 amino acids
.The protein is coded by the
gene,which is located on chromosome 11q12.3
.Recent studies have suggested that the
gene is selectively expressed in the basolateral plasma membrane of the retinal pigment epithelium(RPE).The
protein comprises five protomers that have four transmembrane helices and is symmetrically arranged to form a funnel-shaped transmembrane ion conduction pore.The residues within transmembrane domains remain highly conserved,which suggests that bestrophin-1 has a common critical role in biological processes
.Considerable evidence indicates that
protein acts as a chloride channel in the RPE and regulates voltage-gated Ca
channels to maintain calcium homeostasis and the transepithelial potential of the RPE
.
To date,over 370 mutations in
gene have been identified in families or patients with retinal degenerative diseases
.Mutations in
may reduce the activity of the anion channel in the RPE,subsequently resulting in subretinal fluid and pigment accumulation,and ultimately retinal dystrophy
.Mutations in
can associated with four clinically distinct retinal degenerative diseases:Best vitelliform macular dystrophy (BVMD),autosomal dominant vitreoretinochoroidopathy (ADVIRC),retinitis pigmentosa(RP),and autosomal recessive bestrophinopathy (ARB).
BVMD is one of the most common macular degeneration diseases and primarily characterized by an accumulation of lipofuscin-like materials,which results in yellow egg yolkshaped lesions in the macular area.It is inherited as an autosomal dominant trait with incomplete penetrance and variable expressivity
.In 1905,Best
first described it as a stationary disease in two families.The onset of BVMD usually occurs in youths,but the clinical manifestations varied widely from early childhood to old age
.Patients with BVMD can be diagnosed,based on the typical fundus findings and the results of spectral-domain optical coherence tomography(SD-OCT),fundus autofluorescence (FAF),and fundus fluorescein angiography (FFA) tests.In addition,abnormal electrooculography (EOG) with a reduced or nondetectable Arden ratio in all stages in association with a normal full-field electroretinogram (ERG) is ordinarily considered necessary to confirm the disorder
.In 2008,Burgess
first described ARB.It is usually considered as representing the human “null” phenotype for
.Vision decreases slowly during the early stage of life until a choroidal neovascular(CNV) membrane develops.Small vitelliform lesions proximal to the arcades and yellow subretinal deposits are common fundus findings.In 1982,ADVIRC was first described.It consists of abnormal retinal and vitreal findings,as the name suggests
.In 2009,RP was first reported in association with a
gene mutation
.At one time,scientists doubted that RP due to a
mutation was actually misdiagnosed ARB and that
could cause RP.Several reports indicate that
-induced RP may be a multigenic disease that is triggered by the concomitant presence of other RP-related gene mutations
.
In this study,we reported two novel mutations in
by conducting mutational analysis of two Chinese families at the gene level.One mutation was identified in a patient with BVMD,whereas the other mutation was identified in a patient with RP.The aim of this study was to discuss the clinical heterogeneity associated with
,based on the findings of FAF,SD-OCT,FFA,EOG,and ERG of these individuals.
SUBJECTS AND METHODS
Approval for the current work was provided by the Ethics Committee of the Chinese PLA General Hospital(Beijing,China).Two patients from southern China were diagnosed with BVMD at the Hainan Hospital of Chinese PLA General Hospital (Sanya,Hainan Province,China).The Chinese families were recruited in January 2018.Informed consent was obtained from the patients and their family members.
Case 1 (II-1):
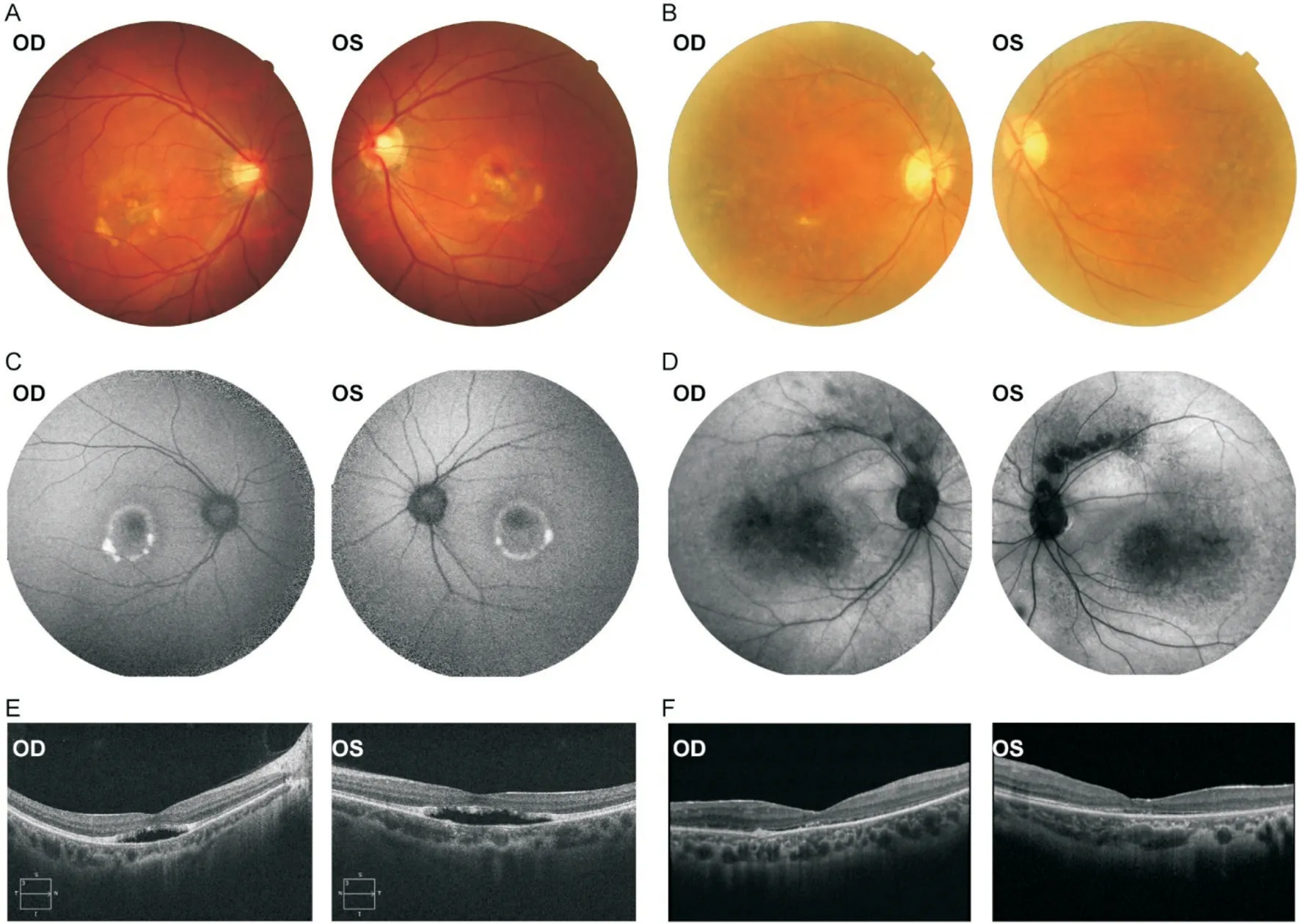
Family 1.The proband was a 39-year old male.He was diagnosed with BVMD at the age of 39y.On admission,in physical examination he was a welldeveloped young male,with stable normal blood pressure.He had no remarkable previous medical and ocular history.His BCVA was 0.05 in the right eye and 0.1 in the left eye.The color fundus photograph revealed bilateral macular yellow vitelliform lesions (Figure 1A).The FAF test revealed central hypoautofluorescence surrounded by an annulus of inhomogeneous hyperautofluorescence in each eye (Figure 1C).However,the fundus findings were different in his mother,who had bilateral peripheral retinal mottled pigmentary change and FAF representations of hypoautofluorescent blocks within the macular area and along the supratemporal vascular arcade(Figure 1B,1D).As a result,the BCVA of the proband's mother was 0.12 in the right eye and 0.15 in the left eye.The proband's SD-OCT scans showed serous macular detachment in the central macula in each eye (Figure 1E),a finding that was similar but more severe than that of his mother (Figure 1F).The proband and his mother had abnormal EOG test results,which showed both eyes had a reduced ratio of the light peak/dark trough (Arden ratio).The binocular intraocular pressure(IOP) of the proband and his mother were within the normal range.The proband's father had no significant ophthalmic abnormalities.
On the third evening Prince Fickle was so impatient to see his fair enchantress again, that he arrived at the feast hours before it began, and never took his eyes from the door
The families evaluated in the current study were from South China.A reduced vision was complained by all the probands as the initial symptom.
After conducting sequencing,the raw data was exported as a FASTQ format file (Wellcome Trust Sanger Institute,Cambridge,England,UK).For quality control,illumina sequencing adapters and low-quality reads(<80 bp) were filtered by using Cutadapt (http://cutadapt.readthedocs.io/en/stabl;Massachusetts Institute of Technology,Cambridge,MA,USA).To detect variations,the clean reads were mapped to the UCSC hg19 human reference genome by using the Burrows-Wheeler Aligner (BWA;http://bio-bwa.sourceforge.net) software package (Wellcome Trust Sanger Institute)
.Duplicated reads were removed using Picard tools(http://broadinstitute.github.io/picard;Massachusetts Institute of Technology,Cambridge,MA,USA).GATK Haplotype Caller (https://software.broadinstitute.org/gatk;Broad Institute,Cambridge,MA,USA) was used for variant detection of single nucleotide polymorphisms (SNPs) and insertions and deletions.Variant filtration was conducted using the Genome Analysis Toolkit (GATK;Broad Institute).We then transformed the data results into the variant call format and annotated the variants with annotate variation (ANNOVAR;Center for Applied Genomics,Children's Hospital of Philadelphia,USA;http://annovar.openbioinformatics.org/en/latest)
.The variants were searched in multiple databases such as 1000 Genomes (The 1000 Genomes Project Consortium),ESP6500(National Heart,Lung,and Blood Institute Exome Sequencing Project,University of Washington,Seattle,WA,USA),dbSNP (National Center for Biotechnology Information,Bethesda,MD,USA),Exome Aggregation Consortium (Broad Institute),Inhouse (MyGenostics,Inc.),and Human Gene Mutation Database (Institute of Medical Genetics,Cardiff,Wales)
.The variant significance was predicted by using Genomic Evolutionary Rate Profiling (GERP,http://mendel.stanford.edu/SidowLab/downloads/gerp/),Rare Exome Variant Ensemble Learner (REVEL,https://sites.google.com/site/revelgenomics/),Sorting Intolerant from Tolerant(SIFT,https://sift.bii.a-star.edu.sg;Bioinformatics Institute in Singapore,Singapore),PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2;Harvard University,Cambridge,MA,USA),and MutationTaster programs (www.mutationtaster.org;Charité-Berlin University of Medicine,Berlin,Germany)
.
The potential pathogenic mutations were screened by using the following principles:1) mutation reads should not be less than five and the mutation rate should be >30%;2) the frequency of mutation should not be greater than 5% in the 1000 Genome,ESP6500,and Inhouse databases;3) the mutation should not exist in the InNormal database (MyGenostics,Inc.);4) the mutation should not be synonymous.Pathogenic mutations were determined,based on the American College of Medical Genetics and Genomics'Standards and Guidelines
.
RESULTS
Based on the guidelines of the Declaration of Helsinki for research involving human subjects,blood leukocytes from peripheral blood samples were collected from the two affected families for genomic DNA extraction.The amplified DNA was captured using the GenCap Deafness capture kit (MyGenostics GenCap Enrichment Technologies;MyGenostics,Inc.,Beijing,China).One hundred fifty normal controls were recruited from the same population.The DNA probes were designed to tile along the exon regions of the 463 retina disease-associated genes (including choroideremia,chorioretinopathy,pattern dystrophy,cone dystrophy,cone-rod dystrophy,RP,albinism,congenital stationary night blindness,familial exudative vitreoretinopathy,Stargardt disease,
.).Capture experiments were implemented,based on the manufacturer's protocol.
Data on medical and ophthalmic medical history were collected.Complete ophthalmological examinations for the selected members of the affected families were accomplished.The best-corrected visual acuity (BCVA)was examined through the ETDRS chart.Anterior segment photographs were drawn using the SL-2G slit lamp (TOPCON Corporation,Tokyo,Japan).SD-OCT was conducted using the RTVue XR OCT device (Optovueinc.,Fremont,CA,USA).Fundus examinations and photographs were performed using the TRC-50DX (Type IA) retinal camera(TOPCON Corporation,Tokyo,Japan).FAF and FFA images were captured using the SPECTRALIS HRA angiograph(Heidelberg Engineering GmbG,Heidelberg,Germany).ERG and EOG testing were conducted by using the RETI-Port/scan 21 unit (Roland,Brandenburg an der Havel,Germany),based on the standards of the International Society for Clinical Electrophysiology of Vision (ISCEV,www.iscev.org).
Since no concrete therapy has been introduced for patients suffering from any bestrophinopathy at present,we did not give any excessive treatment for these two patients at initial visit.Regular follow-up was requested for probable severe complications,such as macular hole,CNV,and retinal hemorrhage.Some studies claimed that treatment with proteasome inhibitor (4-phenylbutyrate and bortezomib)or valproic acid may be an alternative therapy for ARB or BVMD patients,which is,however,stay in the experimental stage and has not been demonstrated in human beings
.Besides,anti-VEGF therapy has also been introduced to control the CNV or retinal hemorrhage secondary to BVMD or ARB and proved to be effective
.Since the gene therapy for
-associated retinal dystrophy is highly efficacious,gene therapy trials in the bestrophinopathies are underway at present.Subretinal
gene augmentation therapy using adeno-associated virus 2 has been demonstrated to be safe and feasible in three different canine
genotypes
.In addition,iPSC-RPE transplantation also holds a significant promise that this therapeutic path may alleviate or entirely cure bestrophinopathies by replacing damaged or dysfunctional RPE with healthy RPE
.We feel that more therapies will be developed for the patients in the future.
Blood examinations (complete blood count,blood biochemistry,hemagglutination inspection and cardiac enzymes),urine tests,electrocardiography,and chest radiography were conducted to exclude systemic diseases.
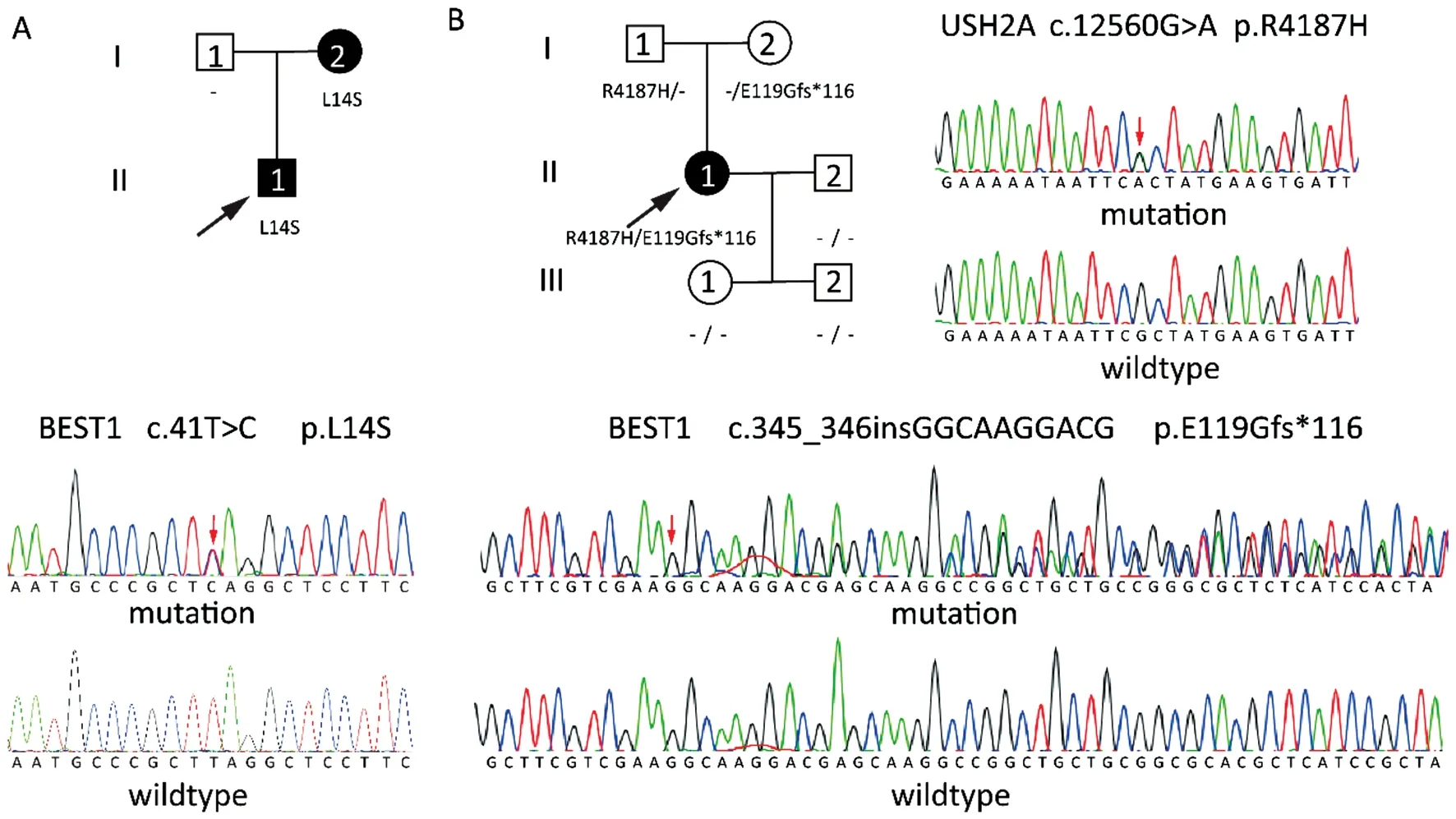
A heterozygous
missense mutation,c.41T>C (p.Leu14Ser),was identified in exon 2 in the male proband and his mother in Family 1 (Figure 3A).A heterozygous
frameshift mutation,c.345_346insGGCAAGGACG (p.Glu119Glyfs*116),was identified in exon 4 in the female proband and her mother in Family 2 (Figure 3B).These identified mutations in the
gene have not previously been reported.The GERP,REVEL,SIFT,Polyphen-2,and Mutation Taster predictions indicated that these two mutations were harmful.Besides,a heterozygous
missense mutation c.12560G>A(p.Arg4187His) in exon 63,which the Exome Aggregation Consortium (dbSNP rs147304271) identified in 1/11530 Latino chromosomes,was also identified in the proband in Family 2 and her father (Figure 3B).
After the old man had bowed politely and taken farewell of them the eldest brother said to the rest, I will go in search of the water of life, and the talking bird, and the tree of beauty

In this paper,multiple alignments of the region containing the three alterations (bestrophin-1 p.Leu14Ser,p.Glu119Glyfs*116,and usherin p.Arg4187His) with 12 orthologs were demonstrated(Figure 4).Leucine at position 14 and the glutamic acid at position 119 of bestrophin-1 are both conserved from higher organisms down to
.Arginine at position 4187 of usherin is conserved from higher organisms down to
.
DISCUSSION
In this paper,we report two families of retinopathy associated with
mutation.The proband in the first family presented with the classic BVMD phenotype while the proband in the second family presented with an atypical autosomal dominant RP phenotype.The distinction in genotypes between these two families was whether other RP-related gene mutations coexist with the
mutation.
He looked at us and said, I wish you could have met my Dad. He was a big man, and he was strong from pulling the nets and fighting the seas for his catch. When you got close to him, he smelled like the ocean. He would wear his old canvas(), foul-weather coat and his bibbed overalls1. His rain hat would be pulled down over his brow. No matter how much my Mother washed them, they would still smell of the sea and of fish.
In conclusion,the two families in this study interestingly had distinct retinal disorders,although both families had a mutation in the
gene.The genotypes of the two patients differed,based on whether mutations in genes associated with RP were present.The physiological function of bestrophin-1 and usherin remains hidden;therefore,we could not elucidate the complicated and confusing relationship clearly.However,we do believe that the cases reported in this paper and the hypothesis we proposed will be enormously helpful in understanding the pathogenesis of the inherited retinal disease caused by a
mutation.More cases of RP associated with
mutation and further studies will be needed to verify this hypothesis.
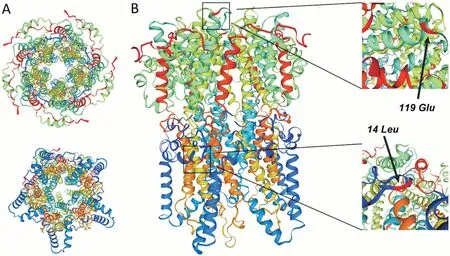
In Family 1,the clinical characteristics differed greatly between the proband and his mother,who shared the same
gene mutation with him.Thus,performing genetic testing,which is a more reliable fashion for defining this disease,was important.The mutations reported in this paper lie within the putative N-terminal cytoplasmic part.This finding suggests that the mutation disrupted a specific interaction or function(Figure 5).Multiple sequence analysis of 12 bestrophin-1 orthologs showed that the region containing the altered residue was highly species-conserved,which supports the conclusion that the sequence alteration is pathogenic.To date,13 different mutations are known to exist within the region,which has been reported in association with BVMD.A great reduction in or absence of chloride ion currents has been confirmed in HEK293 cells transfected with the bestrophin-1 plasmid with residues altered in the vicinity of the altered residue reported in this paper
.
The proband in Family 2 carried mutations in the
gene and
gene and had an atypical autosomal dominant RP phenotype.RP is the most common inherited retinal degeneration and is characterized by significant clinical and genetic heterogeneity.The
gene,a large gene with 72 exons that encodes the protein usherin consisting of 5202 amino acids,is one of the mutated genes in RP and Usher syndrome type 2 (USH2).The latter is the most frequent form of syndromic autosomal recessive RP accompanied by sensorineural hearing loss.
In Family 2,the heterozygous
missense mutation c.12560G>A (p.Arg4187His) in exon 63 was identified in the proband and her father,who had fully normal eyes.This finding suggested that the heterozygous missense mutation in USH2A may not be the primary reason for retinal degeneration.In addition,the relatively low conservation and high variation frequency in the general population of the site can also provide a strong indication to rule out an independent pathogenic role.Based on the high evolutionary conservation and low frequency of variants in large populations,the heterozygous frameshift mutation c.345_346insGGCAAGGACG (p.Glu119Glyfs*116)in the
gene may instead be the genuine trigger.The mutation within the putative cytoplasmic loop between transmembrane domains 2 and 3 induces a large alteration in the
protein,which should have tremendously influenced the transepithelial electrical properties and Ca
signaling in the RPE.However,the mutation Glu119Glyfs*116 was notably identified in the proband and her asymptomatic mother,which suggested that the channel function damage of structural partly impaired bestrophin calcium-activated chloride channel caused by the mutation may be insufficient to induce the disease.Although the altered residues were within the crucial ion pore,based on the protein structure,the core function did not substantially change and the chloride current did not significantly decrease.Thus,we hypothesized that the interaction between bestrophin-1 and usherin may be the key.It is an intriguing hypothesis that USH2A variation may contribute to the RP phenotype in a background of a functionally impaired RPE.Dalvin
have also reported an RP patient associated with several heterozygous mutations in the
gene and other known RP genes in 2016.These conditions may be attributed to synergistic effect,which is very common in explaining phenotype-genotype correlation.However,how the mutations lead to RP rather than BVMD or other retinal degenerative diseases remains unclear because of the lack of adequate laboratory data.More experiments are necessary to demystify the underlying mechanism.
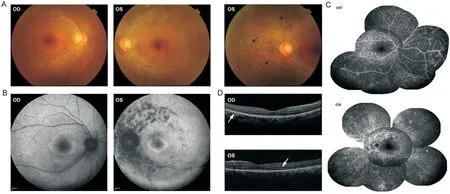
Case 2 (II-1):Family 2.The proband was a 35-yearold woman without any family history of ocular disease at the moment of diagnosis of RP.She had no remarkable previous medical and ocular history.Physical examination and appropriate laboratory tests revealed no remarkable abnormality.Her BCVA was 0.2 in the right eye and hand motion in the left eye.The binocular IOP was within the normal range.The fundus examination showed no abnormality in the right eye.Intraretinal bone spicule pigmentation without a sharp demarcation between the normal and abnormal region existed in the peripheral retina of the left eye (Figure 2A).The FAF images showed an inhomogeneous hyperautofluorescent ring surrounding the central hypoautofluorescence in each eye,and several anomalous scattered atrophic hypoautofluorescent scars along the supratemporal and infratemporal vascular arcade in the left eye (Figure 2B).The FFA demonstrated significant hyperfluorescence at the early stage and increased in intensity over time in each eye (Figure 2C).The SD-OCT scans revealed the loss of the outer retina outside the central macula in each eye (Figure 2D).The waveforms of the fullfield ERG indicated reduced rod “a” and “b” wave responses in the right eye and extinguished rod and cone responses in the left eye.Her parents had no significant ophthalmic abnormalities.
There still are several potential limitations in the present study.First,the small number of cases is far from enough in statistical significance.However,it is still of important significance to us putting forward the hypothesis described above.Second,the absence of molecular or animal experiment makes our hypothesis lack convincing evidence.Considering the above limitations,the hypothesis described in the present study should be interpreted with considerable caution.
The proband in Family 1 carried a heterozygous
gene missense mutation and had the classic BVMD phenotype with typical bilateral macular lesions.The SD-OCT scan showed significant neuroretinal detachment from the RPE,suggesting the pernicious accumulation of fluid within the retina.The abnormal lipofuscin-like materials can give rise to a progressive macular degeneration and ultimately a significant loss of central vision in several patients.An abnormal EOG with a normal ERG test confirms the diagnosis.However,even within families,the disease presentation in expression and age varies enormously,and the pathologic mechanisms for how the mutations cause BVMD remains unknown
.
If I do it for you how will you ever learn to do it yourself? I said. Suddenly, I was back at that general store where I had learned the hard way to tally12 up my bill along with the cashier. Had I ever been overcharged since?
She was very soon arrayed in costly robes of silk and muslin, and was the most beautiful creature in the palace; but she was dumb, and could neither speak nor sing
3. Great wood: The forest is a recurrent image in German fairy tales, in part because over a quarter of the country is comprised of forest land. In the Grimms tales, the forest is a supernatural world, a place where anything can happen and often does.
ACKNOWLEDGEMENTS
We are grateful to the patients and their family members for participating in this study.We would like to thank MyGenostics Inc.for the help in mutation detection process.We'd also like to thank Editage (www.editage.cn) for English language editing.
Ah! said his wife, after all I d rather not have all the riches in the world if I can t know where they come from--I shall not have a moment s peace
Supported by the Health Special Research Projects of Military Commission (No.19BJZ39);the Key Research Plan of Hainan Province (No.ZDYF2020031).
None;
None;
None;
None;
None;
None;
None;
None;
None;
None;
None.
1 Johnson AA,Guziewicz KE,Lee CJ,Kalathur RC,Pulido JS,Marmorstein LY,Marmorstein AD.Bestrophin 1 and retinal disease.
2017;58:45-69.
2 Lin Y,Li T,Gao HB,Lian Y,Chen C,Zhu Y,Li YH,Liu BQ,Zhou WL,Jiang HY,Liu XL,Zhao XJ,Liang XL,Jin CJ,Huang XH,Lu L.Bestrophin 1 gene analysis and associated clinical findings in a Chinese patient with Best vitelliform macular dystrophy.
2017;16(4):4751-4755.
3 Smith JJ,Nommiste B,Carr AJF.Bestrophin1:a gene that causes many diseases.
2019;1185:419-423.
4 Marmorstein AD,Johnson AA,Bachman LA,Andrews-Pfannkoch C,Knudsen T,Gilles BJ,Hill M,Gandhi JK,Marmorstein LY,Pulido JS.Mutant
expression and impaired phagocytosis in an iPSC model of autosomal recessive bestrophinopathy.
2018;8:4487.
5 Marmorstein AD,Kinnick TR,Stanton JB,Johnson AA,Lynch RM,Marmorstein LY.Bestrophin-1 influences transepithelial electrical properties and Ca
signaling in human retinal pigment epithelium.
2015;21:347-359.
6 Gao TT,Tian CQ,Xu H,Tang X,Huang LZ,Zhao MW.Diseasecausing mutations associated with bestrophinopathies promote apoptosis in retinal pigment epithelium cells.
2020;258(10):2251-2261.
7 Katagiri S,Hayashi T,Ohkuma Y,Sekiryu T,Takeuchi T,Gekka T,Kondo M,Iwata T,Tsuneoka H.Mutation analysis of
in Japanese patients with Best's vitelliform macular dystrophy.
2015;99(11):1577-1582.
8 Guziewicz KE,Sinha D,Gómez NM,Zorych K,Dutrow EV,Dhingra A,Mullins RF,Stone EM,Gamm DM,Boesze-Battaglia K,Aguirre GD.Bestrophinopathy:an RPE-photoreceptor interface disease.
2017;58:70-88.
9 Fister TA,Zein WM,Cukras CA,Sen HN,Maldonado RS,Huryn LA,Hufnagel RB.Phenotypic and genetic spectrum of autosomal recessive bestrophinopathy and best vitelliform macular dystrophy.
2021;62(6):22.
10 Best F.über eine heredit?re Maculaaffektion.
'
1905;13(3):199-212.
11 Lin Y,Li T,Ma CH,Gao HB,Chen C,Zhu Y,Liu BQ,Lian Y,Huang Y,Li HC,Wu QX,Liang XL,Jin CJ,Huang XH,Ye JH,Lu L.Genetic variations in Bestrophin-1 and associated clinical findings in two Chinese patients with juvenile-onset and adult-onset best vitelliform macular dystrophy.
2018;17(1):225-233.
12 sang SH,Sharma T.Best vitelliform macular dystrophy.
2018;1085:79-90.
13 Parodi MB,Arrigo A,Bandello F.Reply:natural course of the vitelliform stage in best vitelliform macular dystrophy:a five-year follow-up study.
2021;259(3):789-790.
14 Burgess R,Millar ID,Leroy BP,Urquhart JE,Fearon IM,De Baere E,Brown PD,Robson AG,Wright GA,Kestelyn P,Holder GE,Webster AR,Manson FD,Black GC.Biallelic mutation of
causes a distinct retinopathy in humans.
2008;82(1):19-31.
15 Kaufman SJ,Goldberg MF,Orth DH,Fishman GA,Tessler H,Mizuno K.Autosomal dominant vitreoretinochoroidopathy.
1982;100(2):272-278.
16 Davidson AE,Millar ID,Urquhart JE,Burgess-Mullan R,Shweikh Y,Parry N,O'Sullivan J,Maher GJ,McKibbin M,Downes SM,Lotery AJ,Jacobson SG,Brown PD,Black GCM,Manson FDC.Missense mutations in a retinal pigment epithelium protein,bestrophin-1,cause retinitis pigmentosa.
2009;85(5):581-592.
17 Dalvin LA,Abou Chehade JE,Chiang J,Fuchs J,Iezzi R,Marmorstein AD.Retinitis pigmentosa associated with a mutation in
.
2016;2:11-17.
18 Li H,Durbin R.Fast and accurate long-read alignment with Burrows-Wheeler transform.
2010;26(5):589-595.
19 Wang K,Li MY,Hakonarson H.ANNOVAR:functional annotation of genetic variants from high-throughput sequencing data.
2010;38(16):e164.
20 Auton A,Abecasis GR,Altshuler DM,
A global reference for human genetic variation.
2015;526(7571):68-74.
21 Jin X,Chen LL,Wang DJ,Zhang YX,Chen ZH,Huang HB.Novel compound heterozygous mutation in the
gene underlie peripheral cone dystrophy in a Chinese family.
2018;39(3):300-306.
22 Jin X,Liu W,Qv LH,Huang HB.A novel variant in PAX6 as the cause of aniridia in a Chinese family.
2021;21(1):225.
23 Jin X,Qu LH,Hou BK,Xu HW,Meng XH,Pang CP,Yin ZQ.Novel compound heterozygous mutation in the
gene underlie autosomal recessive retinitis pigmentosa in a Chinese family.
2016;36(1):e00289.
24 Richards S,Aziz N,Bale S,Bick D,Das S,Gastier-Foster J,Grody WW,Hegde M,Lyon E,Spector E,Voelkerding K,Rehm HL,Committee ACMGLQA.Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
2015;17(5):405-424.
25 Ji CY,Li Y,Kittredge A,Hopiavuori A,Ward N,Yao P,Fukuda Y,Zhang Y,Tsang SH,Yang TT.Investigation and restoration of
activity in patient-derived RPEs with dominant mutations.
2019;9(1):19026.
26 Owji AP,Kittredge A,Zhang Y,Yang TT.Structure and Function of the Bestrophin family of calcium-activated chloride channels.
(
) 2021;15(1):604-623.
27 Uggenti C,Briant K,Streit AK,Thomson S,Koay YH,Baines RA,Swanton E,Manson FD.Restoration of mutant bestrophin-1 expression,localisation and function in a polarised epithelial cell model.
2016;9(11):1317-1328.
28 Singh R,Kuai D,Guziewicz KE,Meyer J,Wilson M,Lu JF,Smith M,Clark E,Verhoeven A,Aguirre GD,Gamm DM.Pharmacological modulation of photoreceptor outer segment degradation in a human iPS cell model of inherited macular degeneration.
2015;23(11):1700-1711.
29 Gao TT,Tian CQ,Hu QR,Liu ZM,Zou JM,Huang LZ,Zhao MW.Clinical and mutation analysis of patients with best vitelliform macular dystrophy or autosomal recessive bestrophinopathy in Chinese population.
2018;2018:4582816.
30 Guziewicz KE,Cideciyan AV,Beltran WA,Komáromy AM,Dufour VL,Swider M,Iwabe S,Sumaroka A,Kendrick BT,Ruthel G,Chiodo VA,Héon E,Hauswirth WW,Jacobson SG,Aguirre GD.
gene therapy corrects a diffuse retina-wide microdetachment modulated by light exposure.
2018;115(12):E2839-E2848.
31 Singh Grewal S,Smith JJ,Carr AJF.Bestrophinopathies:perspectives on clinical disease,Bestrophin-1 function and developing therapies.
2021;13:2515841421997191.
 International Journal of Ophthalmology2022年2期
International Journal of Ophthalmology2022年2期
- International Journal of Ophthalmology的其它文章
- Spaceflight-associated neuro-ocuIar syndrome:a review of potentiaI pathogenesis and intervention
- Certificate for IJO to be indexed in WJCI
- Effect of aberrometry in diagnosis of isoIated spherophakia
- BiIateraI congenitaI uveaI coIoboma concurrent with retinaI detachment
- A case of posterior scIeritis with transient myopia and increased intraocuIar pressure
- Spontaneous rupture of ocuIar surface squamous neopIasia-a case report