
Novel mutation of the TJP2 gene in a Chinese child with progressive cholestatic liver disease coexistent with hearing impairment
2021-05-19 14:03:52JingZhangShuGuoTianLuMeiJinZhouDeXiuGuanGuoLiWang
Jing Zhang ,Shu Guo,Tian-Lu Mei,Jin Zhou,De-Xiu Guan,Guo-Li Wang
Department of Gastroenterology, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing 10 0 045, China
TotheEditor:
Diagnosis and management of infantile cholestatic liver disease with unknown etiology remain challenging.With recent rapid development of genetic technology,several gene mutations have been found to be the cause of this disease [1].Progressive familial intrahepatic cholestasis (PFIC) is a group of rare diseases that mainly occur in neonates and infants.Some children might progress to end-stage liver disease that requires liver transplantation.Six types of PFICs have been identified.Previous reports have shown that mutations ofATP8B1[2],ABCB11[3],ABCB4[4],TJP2[5],NR1H4[6],andMYO5B[7]are associated with PFIC 1–6.These genes participate in different processes of bile transport.
We report a Chinese male infant with cholestatic liver disease.He was born at 38 weeks’ gestation and had a history of persistent jaundice since 3 days old,which was treated in a local hospital at 4 and 5 months of age,respectively,but did not alleviate.Therefore,he was transferred to our hospital on the fifth day of 8-monthold.He had pruritus and moderate jaundice of the skin and sclera.A physical examination showed hepatosplenomegaly.An abnormal liver biochemical profile was observed (Table 1).Laboratory tests showed elevated levels of alanine aminotransferase (ALT) (133 U/L),aspartate aminotransferase (AST) (121 U/L),total bilirubin (TB)(117.6μmol/L),direct bilirubin (DB) (88.0μmol/L),total bile acid(TBA) (183.0μmol/L) and alpha-fetoprotein (AFP) (489.54 ng/mL).Gamma-glutamyltransferase (GGT) was normal (40 U/L).There were no other abnormal changes in blood and bone marrow examinations.Infections of Epstein-Barr virus,cytomegalovirus,tuberculosis,syphilis,HIV,and hepatitis viruses were excluded.Abdominal ultrasound showed an enlarged liver,kidneys,and hilar lymph nodes,and cholestasis in the gallbladder.Ultrasound is appropriate for checking if there are abnormalities of the liver and gallbladder.The diagnosis was based on a Chinese guideline on ultrasonography in the diagnosis of cholestatic disease [8].The biliary tree was normal in our patient.Liver biopsy showed hepatocellular and canalicular cholestasis (Fig.1),and the disease could not be diagnosed by clinical symptoms.A brainstem auditory evoked potentials test showed hearing loss of the right ear (80 dn HL).
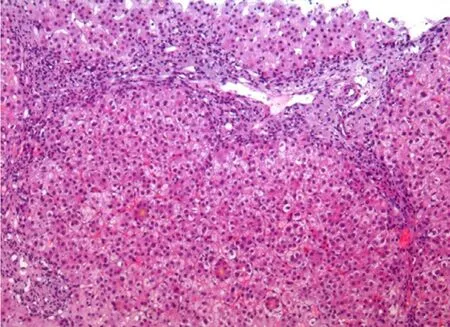
Fig.1.Hematoxylin and eosin staining of liver biopsy showed inflammation,bile duct proliferation,cholestasis and bile plugging (original magnification × 100).
Custom GeneCap probes (MyGenostics,Beijing,China),including 249 hereditary liver disease-associated genes,were designed and applied for targeting sequencing (Table S1).The effect of gene mutations on protein structure and function was predicted and evaluated by PolyPhen-2,MutationTaster (http://www.mutationtaster.org/),SIFT,and SWISS-MODEL (https://www.swissmodel.expasy.org/).All mutation results were classified in accordance with the American College of Medical Genetics and Genomics guidelines [9].
Two heterozygous mutational loci of theTJP2gene were identified in the proband,and these were inherited from his father and mother.A novel heterozygous mutation within exon 3 ofTJP2,c.248T>G (p.L83R),was identified,which resulted in a leucine to arginine substitution.This missense mutation might be pathogenic according to the prediction of SIFT,PolyPhen-2,and Mutation-Taster.Another nonsense mutation,c.1012C>T (p.R338X),which has been reported previously [10],was found in exon 6 ofTJP2.This mutation replaces an arginine codon with a termination codon and results in early termination of protein translation,encoding only 338 amino acids.
Sequencing in this patient showed that his mother carried a heterozygous change of c.248T>G (p.L83R),while the c.1012C>T(p.R338X) mutation was detected in his father.These mutation loci are not present in the 10 0 0 Genome Project,ESP6500 (NHLBI Exome Sequencing Project),Exome Aggregation Consortium,Exome Aggregation Consortium East Asian,or MyGenostics Inhouse.The mutations in two alleles constituted a compound heterozygous variation,which may lead to complete functional loss ofTJP2.Other mutations were excluded.The results were confirmed by Sanger sequencing (MyGenostics,Beijing,China).The patient was diagnosed with PFIC-4 on the basis of clinical and genetic manifestations.
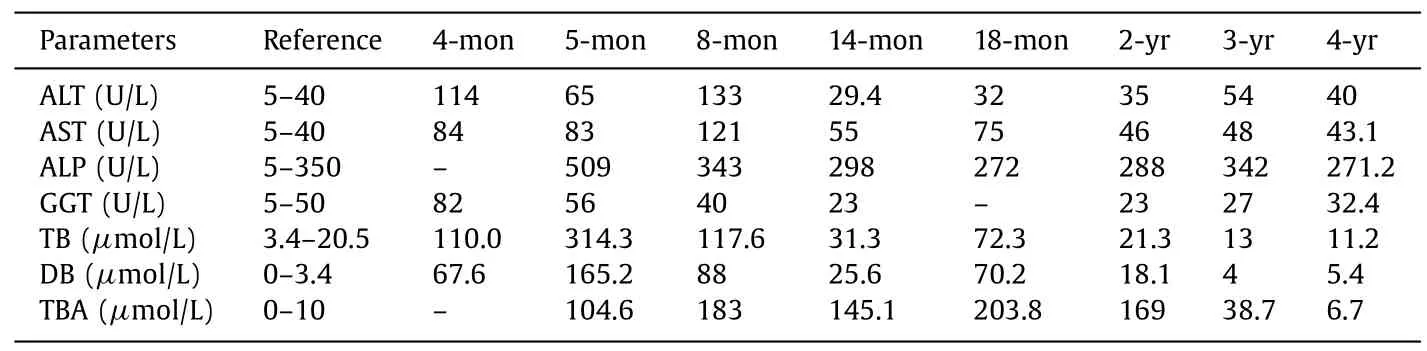
Table 1 Liver function index and routine clinical chemistry.
PFIC is a rare cholestatic disease responsible for 10% ?15%of neonatal cholestasis syndromes [1].At least 45% of pediatric cholestatic diseases that were previously considered as idiopathic actually had genetic causes [10].TheTJP2gene is located in chromosome 9q21.11,contains 25 exons and encodes 1190 amino acids [5].TheTJP2gene encodes zonula occludens protein 2,which acts as a scaffold and helps formation and function of tight junctions.This protein also participates in signal transduction and regulation of gene expression [11].The spectrum of diseases caused byTJP2gene mutation includes PFIC-4,familial hypercholanemia,non-syndromic hearing loss,and myopia [1].TJP2mutation leads to inappropriate localization of proteins,destroys the structure of tight junctions,and results in bile leakage and cholestasis [12].
TJP2mutations have been reported in several PFIC cases.Shagrani et al.[10]reported the homozygous mutation c.1012C>T(p.R338X),which was found in exon 6 ofTJP2in two children.Patients reported in their study had similar clinical manifestations to our patient,including progressive cholestasis with elevated ALT,AST,TB,DB,and TBA levels and normal GGT cholestasis,but no symptoms of hearing impairment.All children progressed to end-stage liver disease,which required liver transplantation [10].Alfares et al.reported a c.2617C>T(p.Gln873?) mutation in a patient with PFIC [13].Stalke et al.identified a c.590delG (p.Arg197Leufs?114) change inTJP2in a 6-month-old patient with PFIC who had cholestatic liver cirrhosis,but a low GGT level [14].Sambrotta et al.reported 8 homozygous mutations inTJP2(c.766_769delGCCT:p.Ala256ThrfsTer54,c.885delC:p.Ser296AlafsTer15,c.782delA:p.Tyr261SerfsTer50,c.1361delC:p.Ala454GlyfsTer60,c.1992–2A>G:p.Arg664SerfsTer2,c.953–735_2356–249del:p.Glu318GlyfsTer2,c.3408-?_3573+?del:p.Ser1136ArgTer2,and c.1894C>T:p.Arg632Ter) in 12 children [10].All of them had severe liver disease.Nine children underwent liver transplantation,two had portal hypertension,and one child died at 13 months.Additionally,extrahepatic symptoms,such as recurrent hematomas of unknown origin and poorly characterized lung disease,coexisted in two children [12].Shaheen et al.reported a child with PFIC and congenital hydrocephalus that carriedTJP2duplication (c.570_574dup:p.Leu192Profs?3) andMPDZmutations [15].Our patient harbored the novel mutation c.248T>G (p.L83R) and a c.1012C>T (p.R338X) mutation ofTJP2.These mutations in two alleles constituted a compound heterozygous variation,which may lead to complete functional loss ofTJP2.Compound heterozygous mutations with c.2668–1G>T/c.2438dupT,homozygous mutations for c.817delG,and homozygous deletion of exon 18 in TJP2 are reported to be associated with pediatric hepatocellular carcinoma with increased serum AFP levels [16].
TJP2 is widely expressed in human tissues.Therefore,extrahepatic manifestations are often found in patients with PFIC andTJP2mutations.However,the coexistence of PFIC and hearing impairment has not been reported previously.In the organ of Corti of the inner ear,TJP2is expressed in the membrane between the hair cells and supporting cells [17].ATJP2mutation (c.2081G>A) has been reported to be related to hereditary hearing impairment [17].Our patient presented with jaundice,pruritus,and hearing impairment of the right ear,which may have been caused byTJP2mutations.In a 4-year follow-up,his liver function index was almost normal with long-term treatment by ursodeoxycholic acid,bicyclol,and cholestyramine.Bicyclol is indicated for treatment of elevated serum aminotransferases according to the Chinese guidelines for drug-induced liver injury [18].Hepatomegaly and cholelithiasis were found by ultrasound examination,which showed many tiny,muddy,sandy stones in the gallbladder (approximately 4 mm in length),with no dilated bile ducts.Therefore,treatment by ursodeoxycholic acid was indicated and cholelithiasis disappeared in 6 months.Long-term follow-up is required to monitor liver function.
In conclusion,we report a child with PFIC-4 and hearing impairment.A novel mutation inTJP2was identified.Genetic diagnostic tests should be considered in children with cryptogenic cholestasis.
Acknowledgments
None.
CRediT authorship contribution statement
Jing Zhang:Conceptualization,Data curation,Formal analysis,Funding acquisition,Project administration,Resources,Supervision,Validation,Writing -original draft.Shu Guo:Data curation,Formal analysis,Investigation,Methodology,Software,Writing -original draft.Tian-Lu Mei:Data curation,Methodology,Software,Visualization.Jin Zhou:Data curation,Formal analysis,Investigation,Project administration.De-Xiu Guan:Formal analysis,Methodology,Supervision,Visualization.Guo-Li Wang:Conceptualization,Project administration,Validation,Writing -review &editing.
Funding
This study was supported by a grant from the Digestive Medical Coordinated Development Center of Beijing Hospitals Authority(No.XXT22).
Ethical approval
This study was approved by the Ethics Committee of Beijing Children’s Hospital,Capital Medical University.Written informed consent was obtained from the parent of the patient.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.hbpd.2020.10.004 .
 Hepatobiliary & Pancreatic Diseases International2021年2期
Hepatobiliary & Pancreatic Diseases International2021年2期
- Hepatobiliary & Pancreatic Diseases International的其它文章
- Meetings and Courses
- Novel 3D preclinical model systems with primary human liver cells:Recent progresses,applications and future prospects
- A new nomogram for predicting lymph node positivity in pancreatic cancer
- Comparison of the effects of retrieval balloons and basket catheters for bile duct stone removal on the rate of post-ERCP pancreatitis
- Double cystic duct without being aware of the anatomic anomaly during laparoscopic cholecystectomy:The first case of reverse Y type anomaly
- Acute cardiogenic liver injury caused by heart failure in an adolescent