
Pitfalls of barcodes in the study of worldwide SARSCoV-2 variation and phylodynamics
2021-03-01 06:52:22JacoboPardo-Seco,AlbertoGómez-Carballa,XabierBello等
Zoological Research 2021年1期
DEAR EDlTOR,
Analysis of SARS-CoV-2 genome variation using a minimal number of selected informative sites conforming a genetic barcode presents several drawbacks.We show that purely mathematical procedures for site selection should be supervised by known phylogeny (i) to ensure that solid tree branches are represented instead of mutational hotspots with poor phylogeographic proprieties, and (ii) to avoid phylogenetic redundancy.We propose a procedure that prevents information redundancy in site selection by considering the cumulative informativeness of previously selected sites (as a proxy for phylogenetic-based criteria).This procedure demonstrates that,for short barcodes (e.g.,11 sites),there are thousands of informative site combinations that improve previous proposals.We also show that barcodes based on worldwide databases inevitably prioritize variants located at the basal nodes of the phylogeny,such that most representative genomes in these ancestral nodes are no longer in circulation. Consequently, coronavirus phylodynamics cannot be properly captured by universal genomic barcodes because most SARS-CoV-2 variation is generated in geographically restricted areas by the continuous introduction of domestic variants.
Analysis of SARS-CoV-2 genetic variation has been widely stimulated by the availability of thousands of coronavirus genomes uploaded to public databases.In particular,the Global Initiative on Sharing all Individual Data (GISAID;https://www.gisaid.org/) offers full open access to SARS-CoV-2 genomic data provided by hundreds of laboratories worldwide.The scientific community can analyze the wholegenome sequences available in these resources to make inferences about SARS-CoV-2 genetic variation and its phylogenetic roots,natural selection,and phylodynamics (Boni et al.,2020; Forster et al.,2020; Gómez-Carballa et al.,2020a,2020b; Gudbjartsson et al.,2020; Rambaut et al.,2020; Rockett et al.,2020; Van Dorp et al.,2020; Yu et al.,2020).Furthermore,the fact that the coronavirus genome is only~30 kb allows for relatively easy computational treatment.
In an attempt to simplify the interpretation of SARS-CoV-2 variation,several recent studies have explored tiny fractions of the genomes by selecting highly informative/variable sites to reconstruct patterns of variation and dispersion of SARS-CoV-2 worldwide using different approaches.These sites together conform a genetic signature or barcode.Zhao et al.(2020)explored informative subtype markers (ISMs) for subtyping SARS-CoV-2 variation to model the geographic distribution and temporal dynamics of COVID-19 spread.Their proposed algorithm identified a compact genetic signature of 11 bp nucleotides (initially 20 bp) in the coronavirus genome,which,according to the authors,defined the most variable (and hence informative) set of sites in these genomes.Similarly,Guan et al.(2020) analyzed a different (but overlapping with Zhao et al.(2020)) signature of 11 nucleotide sites to explore worldwide variation and monitor viral genetic diversity in response to future vaccines or treatments.Instead of using a mathematical algorithm for site selection (as in Zhao et al.(2020)),these authors established their selection on purely phylogenetic criteria.
Based on analysis of >90 K SARS-CoV-2 genomes (herein referred to as the 90 K database) downloaded from GISAID(see Supplementary Data for details),we discuss several issues with these studies that need further consideration,including technical aspects of the methods employed for site selection,and the convenience of monitoring ISMs signatures based on current SARS-CoV-2 phylogeny.In addition,our proposed haplotype entropy (HE) algorithm (see Supplementary Data) corrects the issues of phylogenetic redundancy (Zhao et al.,2020).Most notably,we argue that barcodes provide very limited understanding of the current dispersion patterns of SARS-CoV-2.
Zhao et al.(2020) used entropy to identify “a compact set of nucleotide sites that characterize the most variable (and thus more informative) positions in the viral genomes sequenced from different individuals”.However,there are several issues in the procedure employed by these authors that were not addressed in the publication, which require further consideration.First,their entropy-based algorithm is unable to discriminate variants that are diagnostic of the same phylogenetic branch,basically because these variants have similar frequency in the database (excluding possible phylogenetic homoplasies occurring along the evolutionary history of SARS-CoV-2 genomes) (Figure 1A).To eliminate redundant variants from the initial 20 best ISMs candidates and reduce the list to only 11,the authors examineda posteriorithe evolution of the entropy values over time(entropy covarying over time). We propose that this redundancy could have been eliminated by simply inspecting the SARS-CoV-2 phylogeny.For instance,the phylogenetic tree skeleton in Figure 1A (inspired by Figure 3 in Gómez-Carballa et al.(2020a)),which includes the initial 20 ISMs signature,shows that:variants C8782T–T28144C together define clade B (11 nt compressed ISMs CCTGCCAAGGG in Zhao et al.(2020));the sequence motif C241T–C3037T–A23403G characterizes clade A2 (CC CGCCAGGGG, immediate ancestral node of the most successful SARS-CoV-2 variant outside Asia,which most likely originated in Italy (Gómez-Carballa et al.,2020a)); and G28881A–G28882A–G28883C defines haplogroup A2a4(CCCGCCAGGGA,one of the most important sub-branches of A2 (CCCGCCAGGGG); here we favored the single multinucleotide polymorphism (MNP) event GGG28881AAC for nomenclature,as justified in Gómez-Carballa et al.(2020a)).In addition, their entropy-based algorithm sub-optimally prioritized positions that are diagnostic of nodes located along the same evolutionary pathway,but which add very little to the overall discrimination power of the ISMs set: A1(CCCGCCAAGTG) makes up 4.7% of the total database,while its sub-lineage A1a (CCCTTCAAGTG) represents 4.3%and A1a3 represents 1.8% (Figure 1A).As an alternative,we propose that an algorithm that selects a given ISMs and maximizes the information provided by previously selected ISMs would be more efficient; for example,such a procedure would not include G28882A if it was previously prioritized.Inspired by previous procedures that consider cumulative information provided by sets of genetic variants (Galanter et al.,2012; Pardo-Seco et al.,2014; Salas & Amigo,2010),we propose that an algorithm that exploresHEwould be much more efficient than simply considering individual-site entropy values (see below).
Instead of using a mathematical algorithm,Guan et al.(2020) employed a strict phylogenetic procedure,but their rationale for site selection is also questionable.The authors used genomes from the initial period of the pandemic to reconstruct a phylogenetic tree that derived five major clades(with 15 subclades).Their barcode proposed included a hyper-redundant set of 11 sites where:(i) C8782T–T28144C defined haplogroup B,(ii) set C241T–C3037T–A23403G defined haplogroup A2,while C14408T on top of the A2 sequence motif led to the sublineage A2a, (iii)G1397A–T28688C were both diagnostic of A3a1, and G1440A–G2891A defined haplogroup A4.As in Zhao et al.(2020),these variants defined basal nodes of the SARS-CoV-2 phylogeny,resulting in an unsurprising overlap between their 11 ISMs signatures (Figure 1A).
Another issue in the study of Zhao et al.(2020) relates to the fact that their selection of ISMs was based on entropy values >0.23,and a proportion of “N” and “–“ below 25%; this threshold led the authors to an initial selection of 20 ISMs,but the rationale behind this decision is unsatisfactory.Given the arbitrariness of these thresholds,we are compelled to wonder how much of the total variation has been captured (or,conversely,remains to be explained) by their ISMs barcodes(see below).
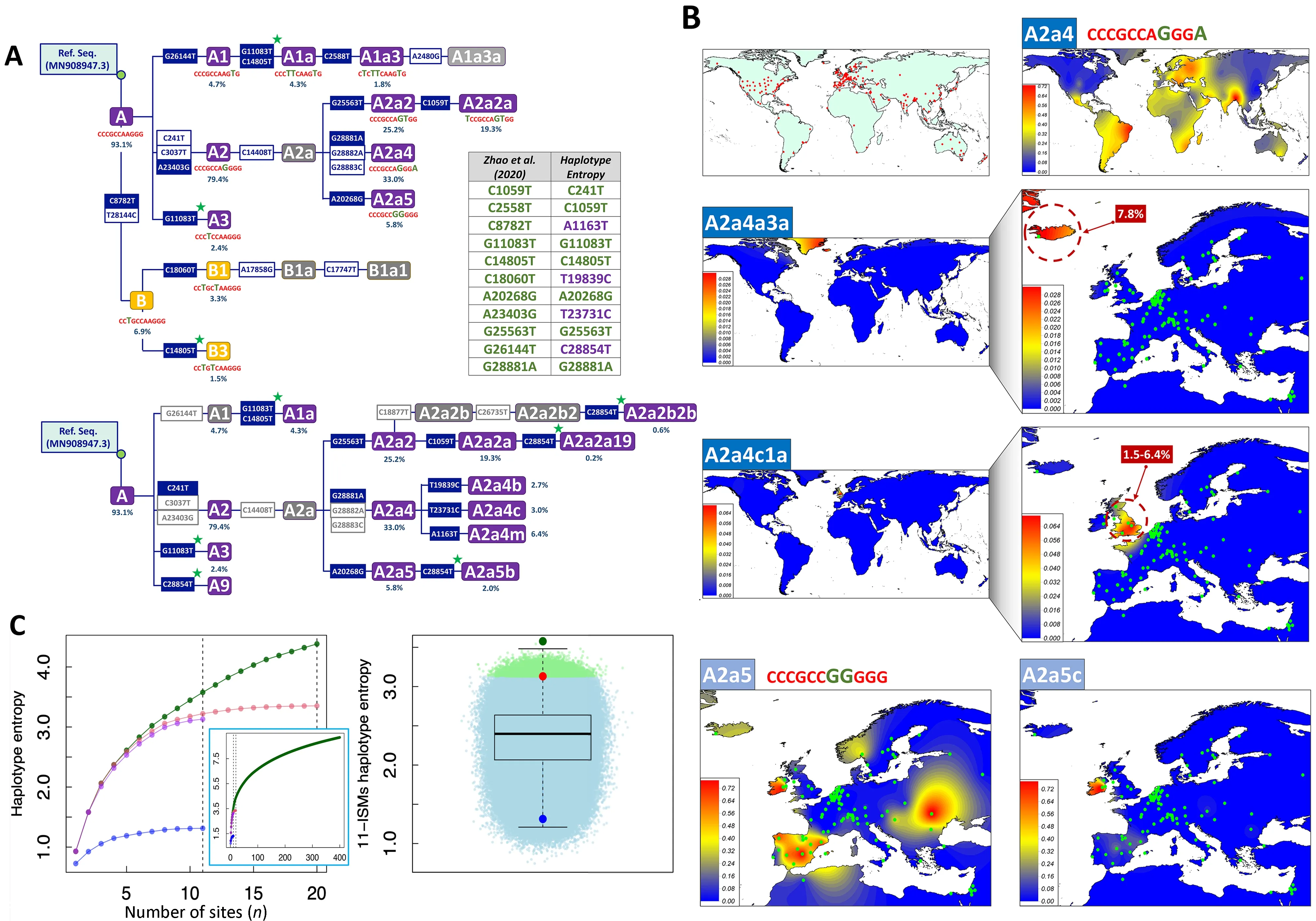
Figure 1 Skeleton of the SARS-CoV-2 phylogeny based on lSMs signatures,interpolated frequency maps of haplogroup sub-lineages having differential geographic distributions,and comparative entropy values for lSMs signatures using different strategies
In addition,Zhao et al.(2020) selected their ISMs using the global GISAID database.This decision conditioned their ability to capture more regional patterns and temporal variations.It is,therefore,not unexpected that their signatures only captured variation located at the basal nodes of the phylogeny. This explains why their algorithm selected diagnostic sites for haplogroups B (CCTGCCAAGGG),B1(CCTGCTAAGGG),A1 (CCCGCCAAGTG),A2 (CCCGCC AGGGG),and A3 (CCCTCCAAGGG) (Figure 1).These basal nodes all occurred at the very initial steps of the pandemic and have spread worldwide (Gómez-Carballa et al.,2020a);therefore,none of the genomes representative of these basal nodes are circulating today,but are only members of derivative phylogenetic branches (note that,according to the evolutionary rate,a mutation accumulates in the SARS-CoV-2 genome approximatelly every two weeks on average (Gómez-Carballa et al.,2020a,2020b)).Moreover,reducing whole SARS-CoV-2 variation to a compact signature of 11 ISMs has an important cost in terms of phylogeographic information.For example,the signature CCCGCCAGGGA (haplogroup A2a4)is widely distributed (>88 countries have representatives of this clade) (Figure 1B).Within A2a4,however,there are sublineages that predominate in different countries; for example,A2a4c1a is almost exclusively present in the UK with frequencies ranging from~1.5% (Wales and Scotland) to 6.4% (England),while the sub-lineage A2a4a3a is exclusively found in Iceland (Reykjavík) at a frequency of 7.8%(Figure 1B).Additionally,the signature CCCGCCGGGGG(haplogroup A2a5) is highly prevalent in Italy,Spain,and Russia based on Figure 5 in Zhao et al.(2020); whereas,according to Gómez-Carballa et al.(2020b),A2a5 most likely originated in Italy and gave rise to one of the most important outbreaks in Spain; furthermore,its sub-lineage A2a5c originated in Spain (most likely in its capital city,Madrid) and its geographic distribution clearly differs from its ancestral node A2a5 (Figure 1B).The information on A2a4a4,A2a4a3a,A2a5c,and many other regionally important clades are all masked by Zhao et al.(2020) under a single signature that corresponds to A2a4 (CCCGCCAGGGA) and A2a5(CCCGCCGGGGG).Moreover,the reductionist view provided by the 11 ISMs barcodes prevents more in-depth interpretation of the principal component analysis (PCA)carried out by Zhao et al.(2020).For example,although this plot does not indicate variation accounted for by PC1 and PC2,it is possible to envisage that the position of the Spanish dataset close to other Asian countries on PC1 is due to the presence of haplogroup B3a (derivative of Asian B3 captured by Zhao et al.(2020) in the signature CCTGCTAAGGG).This haplogroup is much more frequent in Spain than in any other European country (see detailed reconstruction of its origin in Gómez-Carballa et al.(2020b)),a characteristic shared by the USA (and located on the same side of PC1) due to the high frequency presence of B1a1 (captured by Zhao et al.(2020)with the same signature).In addition,the procedure used by Zhao et al.(2020) captured the known D614G spike protein mutation (through their signature CCCGCCAGGGG and derivates; see Figure 1),which is of anecdotal significance only.This amino-acid change corresponds to the RNA change A23403G,which is the most common variant worldwide because it is diagnostic of the basal haplogroup A2 (Figure 1,and Gómez-Carballa et al.(2020a)).The frequency of this variant in Spain is significantly lower (67.2%) than that observed in other European countries (Europe without Spain:85.1%) where haplogroup A derivative clades are much more common.Finally,mutational hotspots constitute an additional problem,because these variants are likely to be included by an algorithm for ISMs selection.This is the case of position 11 083(one of the most important hotspots in the SARS-CoV-2 genome; see Supplementary Material and Supplementary Table S1 in Gómez-Carballa et al.(2020a)).Hotspots are phylogenetically unstable and have poor phylogeographic properties and tracking phylodynamics using hotspots can lead to obscured patterns.
To reduce redundancy in the initial ISMs selection carried out by Zhao et al.(2020),we used theHEalgorithm (see Supplementary Data),which computes the entropy accounted for by haplotypes with an increased number of sites (note that,the 20 and 11 ISMs signatures defined by Zhao et al.(2020)are technically haplotypes).As expected,the computation ofHEprevents the selection of phylogenetically redundant ISMs(because they together define the same phylogenetic branch and/or because of the existence of more complex phylogenetic relationships among variants,e.g.,homoplasies)(Figure 1A).There is an expected overlap between the ISMs selected by theHEalgorithm and those selected by Zhao et al.(2020) because both algorithms tend to select ISMs with the highest individual entropy values and located at the basis of the phylogeny; however,theHEalgorithm significantly improves the overall entropy value compared to that captured by the 11 ISMs signature in Zhao et al.(2020).Thus,when applied to the 90 K database (see Supplementary Data),the entropy of the 11 ISMs signature from Zhao et al.(2020) is 3.1,whereas the 11 ISMs generated by ourHEalgorithm reach 3.6 (i.e.,increase of~16%).When considering profiles uploaded to GISAID until 17 June 2020 (>30 K; high quality genomes),as in Zhao et al.(2020),the total entropy of the 11 ISMs from Zhao et al.(2020) is 3.3,while the 11 ISMs signature based on theHEalgorithm using this dataset leads to a total entropy of 3.5 (increase of~6%).Table 1 shows the entropy values computed for the top ISMs obtained under different scenarios and algorithms; note that theHEalgorithm ranks the 20 ISMs selected by Zhao et al.(2020) in a different way,resulting in a different 11 ISMs signature.Finally,the 11 ISMs barcode proposed by Guan et al.(2020),with a total entropy of 1.3 on the 90 K database,misses the target by far,due to the existing phylogenetic redundancy mentioned above.
TheHEalgorithm leads to a more efficient signature than that captured by the entropy procedure in Zhao et al.(2020);however,none of the algorithms provide information on how far these 11 ISMs signatures are from a hypothetical maximum entropy.To investigate this issue in more detail,we first computed the total entropy of the GISAID 90 K database by considering each genome as a haplotype,obtaining a value of 13.3.This indicates that the 11 ISMs signatures of both Zhao et al.(2020) and theHEalgorithm only capture~27% of the total entropy.We next computed theHEof signatures ranging in size from 1 to 400 ISMs,to see how much information a given ISMs adds to previously incorporated ISMs.Figure 1C indicates that a signature of 400 ISMs captures 9.2 of total entropy; therefore,the remaining 4.1 of entropy in the database (13.3 minus 9.2) is still retained by many other thousands of sites dispersed along the SARSCoV-2 genome.It is worth noting that the incorporation of additional ISMs to a signature adds progressively less and less entropy to the total system (Figure 1C).To further explore the efficiency of the 11 ISMs signature obtained by theHEalgorithm,we computed theHEof the 11 ISMs combined at random from the 50 sites with the highest individual entropy;this “brute force” method eliminated complex phylogenetic relationships that exist between sites (which might not be eliminated by theHEalgorithm).Because combinatorial algorithms are computationally highly demanding (i.e.,>1018possible combinations),we sampled only a reasonable number of combinations (2×106).None of these combinations improved the entropy captured by the 11 ISMs set obtained by theHEalgorithm; however,we observed a total of 12 751 combinations that yielded higher entropy than the signature proposed by Zhao et al.(2020); moreover,>95% of the combinations had higher entropy values than the barcode proposed by Guan et al.(2020) (Figure 1C).By visually exploring the cloud ofHEvalues in these random combinations,it can be inferred that the combination obtained using theHEalgorithm (Figure 1C) is most likely among the best-performing combinations,that is,nearly the topHEpossible with a signature of 11 ISMs.
A final observation of our simulation experiments is that optimal ISMs signatures varied with time.The ISMs set obtained from the global database (from 24 December 2019 to 26 August 2020) differs slightly to the set obtained usinggenomes from the initial phase of the pandemic (from 24 December 2019 to 17 June 2020) and the latest phase in the database (from 17 June to 26 August 2020) (Table 1).
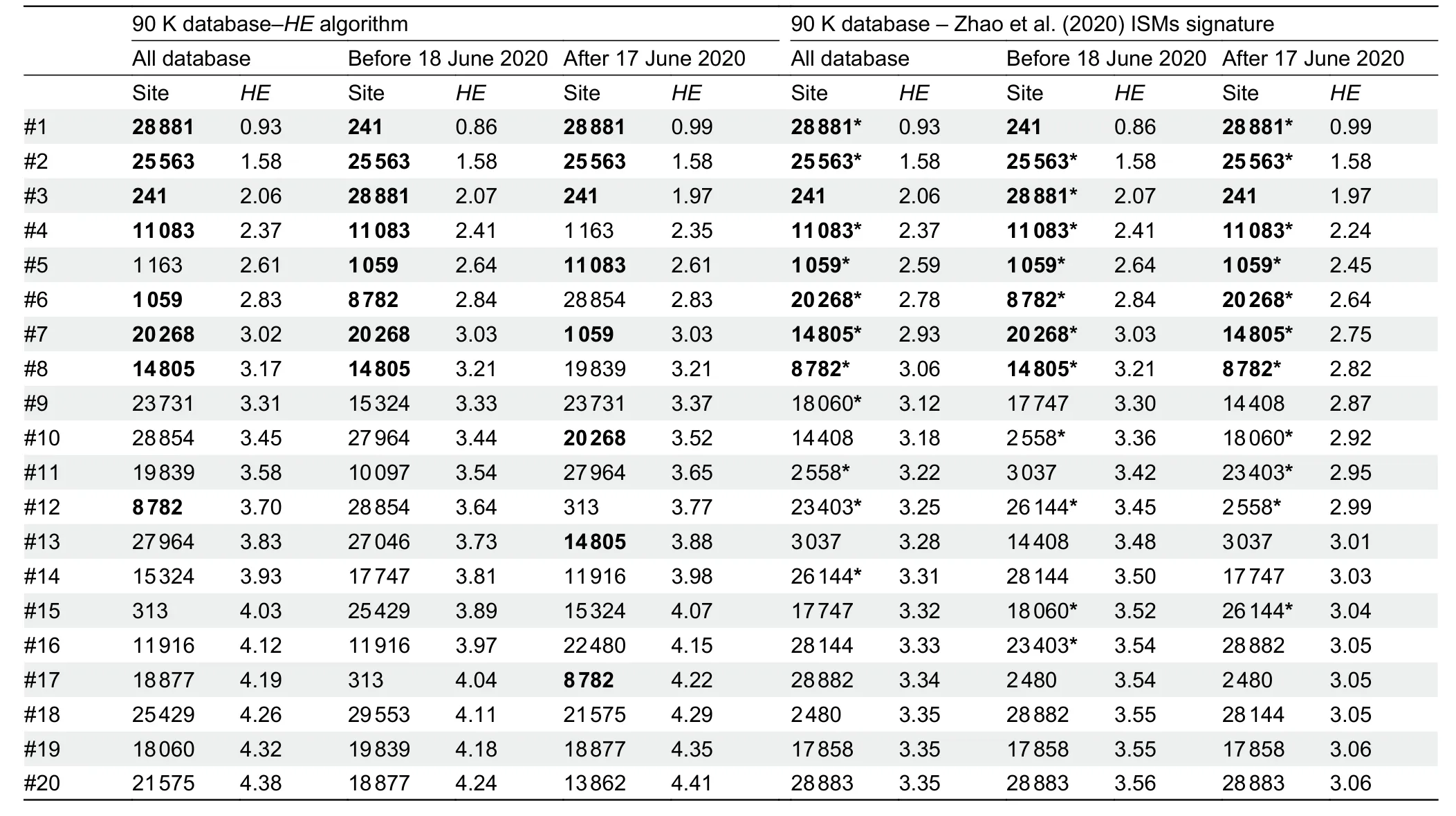
Table 1 lSMs selected using HE procedure described in the present study and 20 lSMs signature captured by Zhao et al.(2020)
As expected,the optimal ISMs set is highly dependent on the variation located at the basal nodes, but optimal signatures can experience small changes depending on the evolution and dispersion of the different SARS-CoV-2 strains worldwide (Table 1).
We have shown that a simple (conceptual) modification to the entropy algorithm employed by Zhao et al.(2020) can lead to a more efficient procedure preventing the selection of sites that have redundant phylogenetic information.Our analysis highlights the need to properly supervise ISMs signatures using known SARS-CoV-2 phylogeny as a more robust approach to shed light on what is really being captured by these signatures.By ignoring phylogeny,the method becomes a kind of ‘black box’ that is difficult to interpret,especially when requiring regional level resolution.The authors attempt to find a parallel between the Nextstrain phylogeny (Hadfield et al.,2018) and their signatures,which does not clarify the sections of the evolutionary tree being captured.Here,we showed (Figure 1A) that a single evolutionarily stable mutational change in the SARS-CoV-2 genome is enough to pinpoint a phylogenetic node in the evolutionary tree.Relatedly,the use of nucleotide strings in the nomenclature of the ISMs signatures represents a major drawback to interpretation and knowledge exchange,rather than an advantage (contraZhao et al. (2020)). Instead, the hierarchical nomenclature used by most scholars (inspired by cladistic theory) appears much more convenient.Guan et al.(2020) proposed a signature based exclusively on phylogenetic criteria.For some unexplained reason,these authors selected highly redundant informative sites and did not realize that their proposal retains only a tiny fraction of global entropy.From here,one can also learn that the use of phylogeny alone does not help to reach the optimal signature,while a strategy that combines mathematical predictions with phylogeny can lead to more appropriate site selection.Most importantly,small ISMs signatures provide a very reductionist view of the pandemic dynamics,which can only superficially inform the story of a few basal phylogenetic nodes(Figure 1A),without accounting for sub-nodes that explain regional patterns and/or arise at different points in time.Regional variation is based on ‘domestic’ mutations that add very little to the global entropy (i.e.,have very limited variation); this variation is,however,very relevant to the region affected in terms of disease spread because it may be responsible for a local/regional outbreak (e.g.,intervention of super-spreader events (Gómez-Carballa et al., 2020a,2020b)).In this regard,the geographic interpretation of signatures in Zhao et al.(2020) seems incomplete and does not really reveal region-specific variation. Briefly, their signature TCCGCCAGTGG (haplogroup A2a2a) is “prevalent in New York and some European countries” but (i) it is even more prevalent in other states of the USA (see their Figure 6)and (ii) it is present in at least 71 countries worldwide (e.g.,USA 48.8%,Israel 54.9%,Denmark 69.7%,Finland 72.9%,Canada 22.3%,Vietnam 21.2%) because it is derived from clade A2a,which earlier originated in Italy (Gómez-Carballa et al.,2020a).Their description around this signature is also confusing,e.g.,position G26144T belongs to a different phylogenetic branch (A1; 50 countries representing all continents); and both A2a2a and A1 emerged from a common ancestor,namely,haplogroup A (Figure 1A).Moreover,their signature CCTGCTAAGGG points to the basal haplogroup B1(Figure 1A; one of the potential phylogenetic roots of the SARS-CoV-2 genome (Gómez-Carballa et al.,2020a)),which is also present in 20 countries at low frequency,except for Canada (19.3%),USA (10.2%),and Mexico (8.5%).As noted by Zhao et al.(2020),it is also highly prevalent in Washington,which is because an ancestral B1 lineage most likely entered the country via early dispersion through the Pacific from Asia,while A2 sub-lineages (e.g.,A2a2a and other A2a derivatives)most likely entered the USA from Europe via the Atlantic side.The concept of a genetic barcode might be attractive for many researchers interested in tracking SARS-CoV-2 variation as a shortcut alternative to whole-genome sequencing.However,as discussed above,future attempts should evaluate the potential limitations of site selection.As demonstrated in the present study,barcodes that capture ancestral SARS-CoV-2 variation may have very limited ability to track recent SARS-CoV-2 dynamics and/or genetic diversity.We envisage that the barcode strategy may be useful to track functional SARS-CoV-2 issues (e.g.,related to virulence,dispersion,vaccine efficiency) that could emerge at any time during the pandemic.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETlNG lNTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRlBUTlONS
A.S.and F.M.-T.conceived the study.A.S.,A.G.-C.,X.B.,and J.P.-S.carried out the phylogenetic and statistical analyses.A.S.prepared the manuscript.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We gratefully acknowledge GISAID and contributing laboratories (Supplementary Table S1) for giving us access to the SAR-CoV-2 genomes used in the present study.

- Zoological Research的其它文章
- The forty-year journey of Zoological Research:advancing with the times
- Chromosomal assembly of the Antarctic toothfish(Dissostichus mawsoni) genome using third-generation DNA sequencing and Hi-C technology
- Geographical range evolution of the genus Polypedates (Anura:Rhacophoridae) from the Oligocene to present
- Molecular and morphological evidence for a new species of the genus Typhlomys (Rodentia:Platacanthomyidae)
- Contribution to the taxonomy of the genus Lycodon H.Boie in Fitzinger,1827 (Reptilia:Squamata:Colubridae) in China,with description of two new species and resurrection and elevation of Dinodon septentrionale chapaense Angel,Bourret,1933
- Dynamic evolution of transposable elements,demographic history,and gene content of paleognathous birds