
Protein–protein docking with interface residue restraints*
2021-01-21 02:16:22HaoLi李豪andShengYouHuang黃勝友
Chinese Physics B 2021年1期
Hao Li(李豪) and Sheng-You Huang(黃勝友)
School of Physics,Huazhong University of Science and Technology,Wuhan 430074,China
Keywords: protein–protein interaction,scoring function,residue restraint,molecular docking
1. Introduction
Proteins are involved in nearly all biological processes in the cell such as DNA replication and repair, RNA transcription, signaling pathways, immune system response, and cell regulation.[1–7]In order to function,almost all of proteins interact with other proteins to form complexes or protein–protein interaction networks.[8,9]Therefore, determining the complex structure of the proteins involved is a crucial step in understanding their interaction mechanism and thus for the development of therapeutic interventions targeting the interactions. However,due to the high cost and technical difficulties,solving the structures of protein complexes by experimental methods is challenging. As such,computational methods such as protein–protein docking become an alternative way to elucidate the complex structure of interacting proteins.[10]Given two individual proteins, protein–protein docking tries to predict the complex structure by sampling putative binding modes of one protein relative to the other and ranking the sampled binding modes according to their binding scores which are calculated by an energy scoring function.[11–17]
In the early stage of the development of protein–protein docking when the number of experimental complex structures is small and information about binding sites is little known,global protein–protein docking, which often refers to ab initio or free docking, is needed to sample possible binding modes over all six degrees (three translational + three rotational)of freedom.[18–35]Although global docking can search entire orientational space for near-native binding modes,most of the binding modes are far from the native structures.Therefore, global docking may result in a higher number of false positives. If information about residue–residue contacts between proteins are available, false positives that give high energy scores can be excluded according to the residue constraints.[36–40]
In fact,such information about binding is common in experiments. For example, the residue constraints can be translated from the NMR experiments.[41]If the experimental information about residue restraints is not available, it can also be derived from the sequences, as the sequences of proteins are directly related to their three-dimensional structures and functions. With the database of sequences growing exponentially,information about the residue contacts between proteins may also be derived through an evolutionary analysis from sequences[42]or deep learning.[43]
To make use of residue contacts obtained from experimental information or mined from sequences,an efficient and fast approach for incorporating interface residue restraints into protein–protein docking has been proposed. The new docking approach, which is named as HDOCKsite, significantly improves the ability of our docking algorithm in distinguishing near-native binding modes from false positives. With the development of residue contact prediction,it becomes more and more valuable to consider residue restraints in ab initio docking. Combining with a good inter-protein contact prediction program,our hierarchical docking program HDOCKsite is expected to achieve accurate docking predictions.
2. Materials and methods
2.1. FFT-based docking algorithm
A fast Fourier transform (FFT)-based hierarchical docking program developed by our group,referred to as HDOCKsite, is used to globally sample putative binding modes, in which an improved shape-based pairwise scoring function has been adopted.[44–46]The advantage of our scoring function is that it takes into account the influence not only from its nearest neighboring receptor grids but also from other farther receptor girds in the form of exp(-1/r2), in which r is the distance between the ligand grid and the receptor grids. Shape complementarity is the primary component of a scoring function and plays a crucial role in protein–protein docking. Despite the significant progresses in current shape complementarity functions, all of them just simply consider the effects of neighboring atoms for a grid point. However, some shape-based interactions such as van der Waals interactions involve not only the nearest-neighboring atoms,but also many more other farther interactions. To solve this problem,we have presented a new pairwise scoring function for our FFT-based docking algorithm to consider the long range effect of protein atoms by an exponential form,which is named as LSC.More details of our improved shape-based pairwise scoring function can be found in our previous study.[44]During global sampling, we use an angle interval of 15?to loop the entire rotational space,and a spacing of 1.2 ?A to search the whole translational space.The search for the translational space is accelerated by an FFT algorithm. For each rotation,the top 10 translations with the best shape complementarities from the FFT-based translational search are optimized by our iterative knowledge-based scoring function ITScorePP.[47–50]The best scored translation is kept for each rotation. Given the angle interval of 15?,there are 4392 evenly distributed rotations in the rotational space.Therefore,we get a total of 4392 binding modes for a protein–protein docking. The ranked binding modes are clustered with a ligand RMSD cutoff of 5 ?A,where the RMSD is calculated only using the backbone atoms of the ligand. If two binding modes have a ligand RMSD smaller than 5 ?A, the one with the worse score is excluded. The docking algorithm has been slightly modified to incorporate residue restraints as described below. A flowchart of our docking protocol is shown in Fig.1.
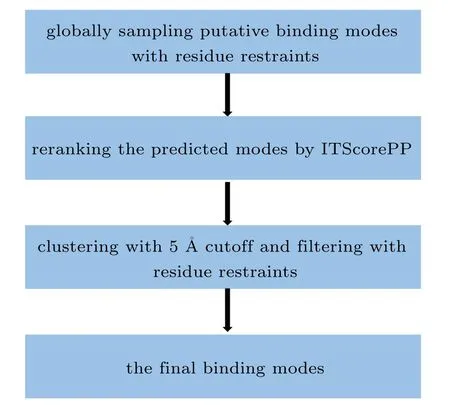
Fig.1. Flowchart of our docking protocol.
2.2. Incorporating residue restraints
The residue restraints derived from the bound structures are incorporated into the FFT-based searching precess and post-docking stage in our hierarchical docking algorithm.During the search process, the residue restraints are mapped onto grids to reduce the computational cost. Specifically, a weight of 100 is assigned to those grid points occupied by receptor or ligand interface residues, and a weight of 10 is assigned to those grid points occupied by receptor-ligand contact residues. The selection of 10 and 100 is based on our scoring function LSC. In LSC, we assign 10 to the imaginary part of the grid located in the protein core region. When two grid points in the protein core region overlap, there is a penalty of 100 for multiplying the imaginary part.[44]During the post-docking stage, only a few thousand binding modes are reserved, and the computational efficiency is no more an issue. Thus,the residue restraints are directly incorporated by examining the distance between the contact residues from the receptor and ligand protein in this stage. The distance cutoff for filtering is initially defined as 8 ?A.If the number of binding modes that satisfy these restraints is less than the specified number of the output binding modes, the distance cutoff will be increased by 1 ?A at a time until enough number of binding modes are output. The maximum number of output binding modes can be designated by the users on the command line,though the default value is 100.
2.3. Definition of contact residues
In the field of inter-protein contact prediction,[42,43,51]two residues in a protein–protein complex are considered contact residues if(i)they belong to two different protein chains and(ii) the minimum distance between their heavy atoms is less than 6 ?A.However,in the CAPRI evaluation protocol,[52–58]a pair of residues at the interface are considered to be in contact if any two of their atoms were within 5 ?A.
Given the fact that two residues within 5.0 ?A or 6.0 ?A are not necessarily in physical contact, we defined the contact residues by calculating the solvent accessible surface area(SASA)of each residue in the bound structures before and after forming the complexes. If the SASA of a residue decreases after forming the complex,it will be considered to be an interface residue.The interface residues on the receptor are defined as the receptor contact residues,and the interface residues on the ligand are considered as the ligand contact residues. If the minimum distance between a receptor contact residue and a ligand contact residue is less than 5 ?A,the residue pair is considered as a receptor-ligand residue contact.
With the definition of contact residues, the bound structures of protein–protein benchmark 4.0 were used to derive the contact residues. With the bound structures aligned with the unbound structures,the corresponding residue ID and chain ID of the unbound structures were output. If there were no corresponding residues in the unbound structures,the residues were excluded. The internal residues whose SASA is smaller than 1 ?A2were also excluded. For each target, we calculated the number of receptor contact residues, ligand contact residues,and receptor-ligand contact residues. As a comparison, we also calculated these contact residues using the above two definitions.
It can be seen from Table 1, the average number of receptor and ligand contact residues by our definition is between the definition of distance cutoff of 5 ?A and the definition of distance cutoff of 6 ?A. This means that the contact residues from our SASA criterion correspond to those for the distance cutoff between 5 ?A and 6 ?A.Our preliminary test also showed that there is no significant difference for different definitions of residue contact.

Table 1. The maximum, minimum, and average number of receptor contact residues, ligand contact residues, and receptor-ligand pairwise contact residues under different definitions for all 176 targets.
2.4. Benchmark data set
The protein–protein docking benchmark 4.0[59]was adopted to evaluate the performance of our hierarchical docking program with different types of residue restraints. There are a total of 176 diverse targets in the benchmark,which contains 123 easy cases, 29 medium-difficulty cases, and 24 difficult cases. The difficulty level reflects their flexibility and is categorized by the root mean square deviation(RMSD)of the interface Cα atoms after superposing the bound and unbound structures. If the interface RMSD between the unbound and bound structures is less than 1 ?A,the cases are considered as easy ones. If the RMSD is between 1 ?A and 2.5 ?A,the cases are regarded as medium difficulty ones.If the RMSD is greater than 2.5 ?A,the cases are deemed as difficult ones.
Each target consists of the co-crystalized bound structures and their related unbound structures for both the receptor and the ligand proteins.The benchmark has been comprehensively used to evaluate the performances of various docking algorithms and scoring functions.[10]In this study, the unbound structures were used for docking, and the native bound complexes were used as the reference during the evaluations.
2.5. Evaluation criteria
The widely used CAPRI evaluation criterion[52–58]was adopted to assess the quality of the predicted binding poses.The evaluation criterion includes three metrics: (i) fnat,the ratio of the native contacts in a predicted binding mode, where a pair of residues from different proteins are deemed to be in contact if any two of their heavy atoms were within 5.0 ?A,to the total number of contacts in the native complex; (ii)Lrmsd,the backbone RMSD of the ligand in the predicted versus target complexes after the receptor proteins are optimally superimposed; (iii)Irmsd,the interface RMSD after the optimal superimposition of the bound and unbound complexes,where a residue belongs to the interface if any of its atoms is within 10 ?A from it interacting protein in the native complex.
Unless otherwise specified, the acceptable quality, in which Lrmsd≤10.0 ?A or Irmsd≤4.0 ?A and 0.1 ≤fnat≤0.3 or fnat≥0.3 and Lrmsd>5.0 ?A and Irmsd>2.0 ?A,was used as a criterion to define a successful prediction. The success rate was used to evaluated the performance of our docking algorithm in binding mode predictions, which was defined as the percentage of the test cases with at least one successful prediction when a certain number of top predictions were considered.
3. Results and discussion
3.1. Overall performance
We have evaluated our hierarchical docking algorithm HDOCKsite with different types of restrains and different numbers of restraints on the unbound structures benchmark 4.0 of 176 targets. Table 2 lists the success rates of our docking algorithm with different types and numbers of restraints in binding mode predictions on the benchmark 4.0 for unbound docking when top 1, 10, 100 predictions were considered. Figure 2 also lists the corresponding success rates when the top 10 predictions were considered. For comparison, Table 2 and Fig.2 also give the corresponding results of ZDOCK 2.3.2. During the docking calculations with HDOCKsite, the receptor restraints,ligand restraints,and receptor-ligand pairwise restraints were randomly selected from the derived receptor contact residues,ligand contact residues,and receptorligand pairwise contact residues, respectively. To reduce the impact of randomness, we randomly selected three sets of residue restraints and repeated the docking calculations with these residue restraints. The average of the results is regarded as the final result.
It can be seen from Table 2 that HDOCKsite with restraints yielded significantly higher success rates than HDOCKsite and ZDOCK2.3.2 without restraints, suggesting the critical role of the residue restraints in docking. HDOCKsite with pairwise restraints obtains a significantly better performance than HDOCKsite with receptor restraints or ligand restraints, and achieves a success rate of 76.7%, 88.4%, and 91.5%for one,two,and three restraints when the top 10 predictions were considered, in comparison to 37.7%, 48.9%,and 53.2% for receptor restraints, and 30.8%, 40.7%, and 45.1% for ligand restraints (Table 2 and Fig. 2). This is because receptor restraints and ligand restraints only constraint the movement of ligand/receptor on the surface of the receptor/ligand. This makes the ligand/receptor lose some of its translational degrees of freedom.Pairwise restraints constraint the distance between the two residues from the receptor and ligand,which makes ligand and receptor lose all of their translational degrees of freedom. Therefore, receptor-ligand pairwise restraints can exclude many more false positive structures that do not satisfy the constraint than receptor restraints and ligand restraints.

Table 2. The success rates (%) of unbound docking with different numbers of restraints predicted by HDOCKsite with receptor restraints,ligand restraints, and receptor–ligand pairwise restraints and ZDOCK2.3.2 on the benchmark 4.0 when top 1, 10, and 100 predictions were considered.
From Table 2 and Fig.2,one can also see that HDOCKsite with receptor restraints achieved a slightly better performance than ligand restraints. This is because the receptors are often larger and thus have a larger surface than ligands.Therefore, a residue restraint designated for the receptor can reduce more search space than a residue restraint designated for the ligand. Namely, the proportion of the designated receptor contact residues not on a ligand surface is greater than the proportion of the designated ligand contact residues not on a receptor surface. Those false positive structures whose designated contact residues do not meet the restraints are excluded. As such, receptor restraints can exclude more false positive structures than ligand restraints.
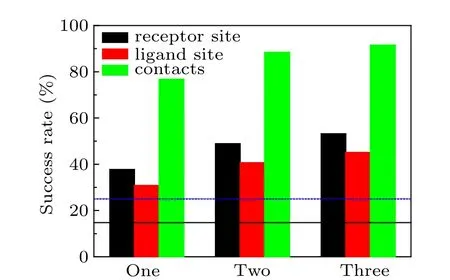
Fig. 2. The success rates of unbound docking for top 10 predictions by our HDOCKsite with different types and numbers of restraints and ZDOCK2.3.2 on the protein–protein docking benchmark 4.0. The dashed line stands for the success rate of ab initio docking without using residue restraint. The solid line represents the success rate of ZDOCK2.3.2 without residue restraints.
Table 2 also shows that HDOCKsite with pairwise restraints achieved an increase of 30.7%in the success rate from zero restraint to one restraint and an increase of 28.5%in the success rate from one restraint to two restraints for the top 1 prediction. However,it only obtained an increase of 12.0%in the success rate from two restraints to three restraints, which is significantly lower than 30.7%and 28.5%. This is because one restraint constraints the relative translation between the receptor and ligand. False positive structures that do not satisfy these translation constraints are excluded, which greatly improves the success rate. Two restraints not only constraint the relative translation but also limit the relative rotation, which will significantly reduce the search space and thus improve the success rate. Since two restraints have reduced most of the search space for possible binding modes, it is expected that three restraints will no longer be able to obviously improve the docking performance.
In addition,one can also notice from Table 2 that the success rates for different numbers of pairwise restraints are very close when the top 100 predictions are considered, and give 91.1%,93.8%,and 94.2%for one restraint,two restraints,and three restraints,respectively. This is because HDOCKsite has reached the limit of success rates with only one pairwise restraint when the top 100 predictions were considered. The number of residue restraints is not a key factor affecting the success rate any more.For those failed cases,most of them are due to the conformational changes,which significantly change the distance between the pairwise residues.
3.2. Performance by the difficulty types of test cases
In order to study the impact of conformational changes on our docking results with different types and numbers of restraints,we have analyzed the docking results by the difficulty types of test cases. Table 3 lists the success rates for the cases with different difficulty levels predicted by our HDOCKsite with different types and numbers of restraints on the benchmark 4.0 when top 1, 10, and 100 predictions are considered for unbound docking. As a comparison,Fig.3 also shows the corresponding success rates for difficulty types of test cases when the top 10 predictions are considered.
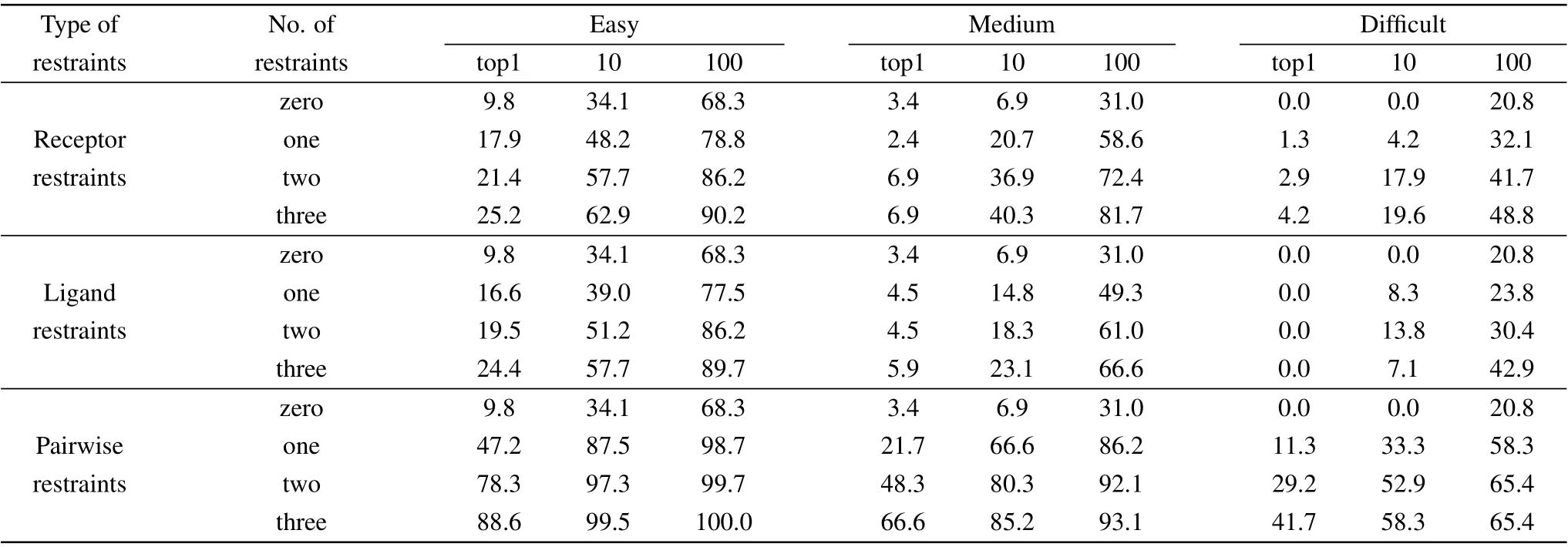
Table 3. The success rates (%) of unbound docking for different difficulties of cases predicted by HDOCKsite with different numbers of receptor restraints,ligand restraints,and pairwise restraints on benchmark 4.0 when top 1,10,and 100 predictions were considered.
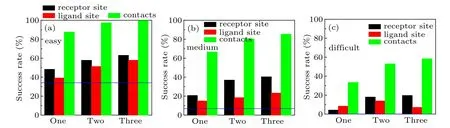
Fig.3. The success rates of unbound docking for top 10 predictions by our HDOCKsite with different types and numbers of restraints on easy(a),medium-difficult(b),and difficult(c)targets of protein–protein docking benchmark 4.0. The dashed line stands for the success rate of ab initio docking without using residue restraint.
When comparing the success rates with different difficulty levels one can find that the conformational changes have a huge impact on the success rate (Table 3 and Fig. 3). The success rates of HDOCKsite for easy cases are significantly higher than those for more difficult cases. For example, easy cases give a success rate of 48.2%, 39.0%, and 87.5% for one receptor, ligand, and pair restraint when the 10 predictions are considered,respectively,compared to 20.7%,14.8%,and 66.6% for medium-difficulty cases and 4.2%, 8.3%, and 33.3%for difficult cases(Fig.3).
Table 3 also shows that for the top 100 predictions with pairwise restraints,HDOCKsite showed different trends in the success rate. For easy cases,HDOCKsite performed similarly for one or more restraints and gave a success rate of 98.7%,99.7%, and 100% for one, two, and three restraints, respectively. For medium-difficulty cases, HDOCKsite gave a success rate of 92.1%and 93.1%for two and three restraints,respectively, which is significantly higher than 86.2% for one restraint. For difficult cases, HDOCKsite gave the same success rates of 65.4%for two and three restraints,which is significantly higher than 58.3% for one restraint. These results indicate that for docking with distance restraints,one restraint is enough for easy cases,while two restraints are needed when the top 100 predictions were considered. This may be understood because ab initio docking has generated many highquality binding modes for easy cases and therefore one distance restraint is enough to rank correct binding modes within the top 100 predictions. However, for medium-difficult and difficult cases, more distance restraints are required to give correct binding modes in the top 100 predictions due to relatively large conformational changes in the unbound structures.
One notable feature in Fig. 3 is that for difficult cases,the success rate of HDOCKsite with three receptor restraints(7.1%)is lower than that with two receptor restraints(13.8%)for top 10 predictions. This is because there are only 24 targets for difficult cases, and therefore the impact of fluctuations is relatively large. In addition,receptor contact residues for difficult cases are expected to experience large conformational changes during the formation of complexes, which would make these receptor restraints no longer valid. Once these invalid restraints are used for docking,the binding modes may be driven to wrong places that meet those invalid distance restraints. This may explain why HDOCKsite with more restraints performed worse than fewer restraints.
3.3. Performance by the quality of predicted binding modes
To study the impact of different evaluation criteria on our docking results with different types and numbers of restraints,Table 4 shows the success rates for different evaluation criteria predicted by our HDOCKsite with different types and different numbers of restraints on the benchmark 4.0 when top 1,10, and 100 predictions were considered for unbound docking. The corresponding results for the top 10 predictions are shown in Fig.4.
It can be seen from Table 4 and Fig.4 that the types and numbers of restraints have little effect on the success rate by the high quality evaluation criterion. This is because the high quality,in which Lrmsd≤1.0 ?A or Irmsd≤1.0 ?A and fnat≥0.5,has exceeded the accuracy limit of the residue restraints. One can also find that the improvements in the success rate by the medium-quality criterion are larger than those by the acceptable and high quality criteria.This may be understood because the distance cutoff of 5–6 ?A for defining residue restraints in this study is consistent with the ligand RMSD cutoff of 5 ?A for the medium-quality criterion(Fig.4).
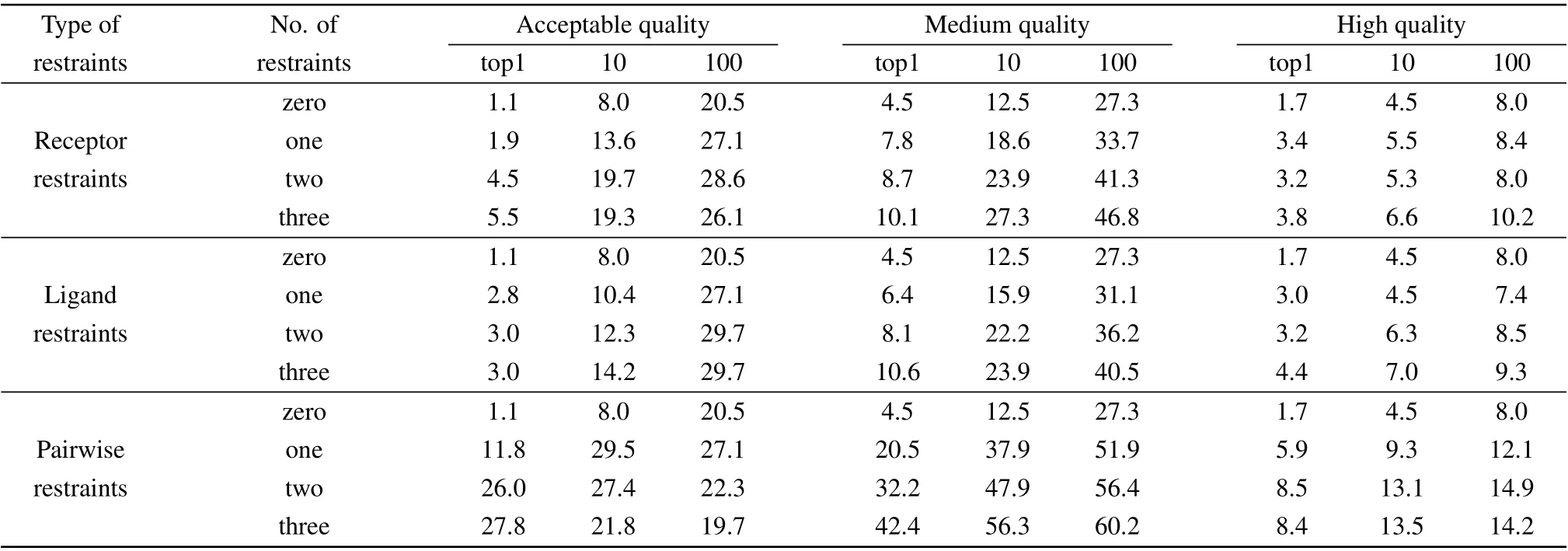
Table 4. The success rates (%) of unbound docking using different evaluation criteria by HDOCKsite with different numbers of receptor restraints,ligand restraints,and pairwise restraints on benchmark 4.0 when top 1,10,and 100 predictions were considered.
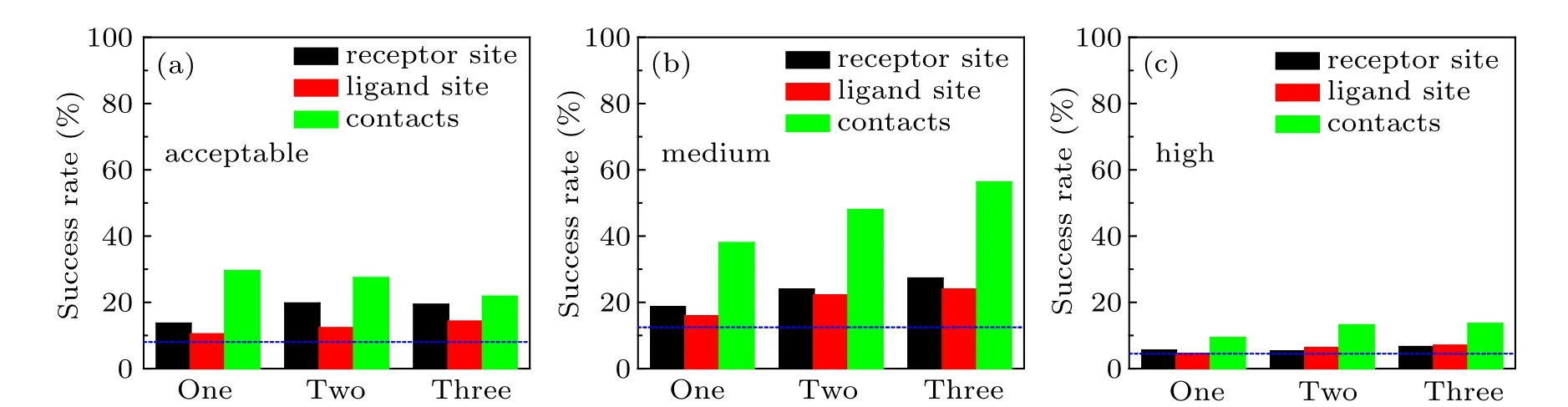
Fig. 4. The success rates of unbound docking for top 10 predictions within the acceptable (a), medium (b), and high (c) accuracies by our HDOCKsite with different types and numbers of restraints on the protein–protein docking benchmark 4.0. The dashed line stands for the success rate of ab initio docking without using residue restraint.
Figure 4 also shows that compared with the cases using acceptable and medium-quality criteria, HDOCKsite with receptor or ligand restraints did not significantly improve the docking performances over the original ab initio docking program using the high-quality criterion when the top 10 predictions were considered. For example, HDOCKsite with one receptor and ligand restraint gave a success rate of 5.5% and 4.5% for top 10 predictions using the high-quality criterion,compared with 4.5% for the original ab initio docking. This may be understood because high-quality binding modes often involve little conformational change and are easy to be identified by ab initio docking.
3.4. Examples of the docking models
Figure 5 shows the top binding modes predicted by our HDOCKsite with different types of restraints for unbound docking on three example targets. As a comparison, the top binding modes by the original ab initio docking are also shown in the figure. For target 2SIC (Fig. 5(a)), the original ab initio docking gave a wrong binding mode with a large ligand RMSD of 53.503 ?A, while HDOCKsite achieved a mediumquality binding mode of Lrmsd=5.731 ?A, Irmsd=1.204 ?A,and fnat= 71.831% with the guide of one residue restraint on the receptor(residue 154:B).For target 1MAH(Fig.5(b)),the original docking program failed to predict a correct binding mode and the top binding mode has a ligand RMSD of 49.096 ?A, while HDOCKsite gave a high-accuracy binding mode of Lrmsd=1.937 ?A,Irmsd=0.905 ?A,and fnat=80.556%with the guide of one residue restraint on the ligand (residue 8:B). For target 1GCQ (Fig. 5(c)), the original docking predicted a wrong binding mode with a large ligand RMSD of 18.737 ?A, while HDOCKsite gave a medium-quality binding mode of Lrmsd=1.960 ?A,Irmsd=1.090 ?A,and fnat=75.556%with one pair of residue restraint from the receptor (residue 210:B) and ligand (residue 595:B). These results suggest the advantage of incorporating residue restraints and the accuracy of our HDOCKsite.
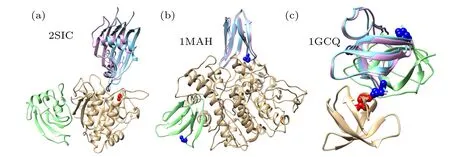
Fig.5.Comparison between the top predicted binding modes before(green)and after(purple)incorporating residue restraints for three example targets,where the native binding mode is represented by the unbound receptor(gold)and ligand(cyan)structures superimposed onto the crystal structure. (a)2SIC with one receptor residue restraint(154:B,red). (b)1MAH with one ligand residue restraint(8:B,blue). (c)1GCQ with one pair of residue restraint(210:B for receptor and 595:B for ligand,red and blue).
4. Conclusion
We have developed an efficient approach to incorporate interface residue restraints into our FFT-based hierarchical protein–protein docking algorithm, which is named as HDOCKsite. We made full use of the restraints by incorporating residue restraints into both searching process and postdocking stage. Our HDOCKsite program was extensively tested on the protein–protein docking benchmark 4.0. It was shown that HDOCKsite significantly improved the docking performances for all types of residue restraints. With only one receptor,ligand,and pairwise restraints,HDOCKsite was able to achieve a success rate of 37.7%, 30.8%, and 76.7% when the top 10 predictions were considered,respectively,compared with 25% for the original ab initio docking. With more and more experimental interface information available, HDOCKsite is expected to be valuable for the development of integrative protein–protein docking. HDOCKsite has been integrated into our web server HDOCK at http://hdock.phys.hust.edu.cn/.

- Chinese Physics B的其它文章
- Numerical simulation on ionic wind in circular channels*
- Interaction properties of solitons for a couple of nonlinear evolution equations
- Enhancement of multiatom non-classical correlations and quantum state transfer in atom–cavity–fiber system*
- Effect of interaction between loop bases and ions on stability of G-quadruplex DNA*
- Retrieval of multiple scattering contrast from x-ray analyzer-based imaging*
- Numerical research on effect of overlap ratio on thermal-stress behaviors of the high-speed laser cladding coating*