
C-reactive protein aggravates myocardial ischemia/reperfusion injury through activation of extracellular-signal-regulated kinase 1/2
2018-09-22 08:02:42WeiNaPEIHaiJuanHUFanLIUBingXIAOYaBeiZUOWeiCUI
Journal of Geriatric Cardiology 2018年7期
Wei–Na PEI, Hai–Juan HU, Fan LIU, Bing XIAO, Ya–Bei ZUO, Wei CUI
?
C-reactive protein aggravates myocardial ischemia/reperfusion injury through activation of extracellular-signal-regulated kinase 1/2
Wei–Na PEI, Hai–Juan HU, Fan LIU, Bing XIAO, Ya–Bei ZUO, Wei CUI
Department of Cardiology, Second Hospital of Hebei Medical Universityand Institute of Cardiocerebrovascular Disease of Hebei Province, Shijiazhuang, Hebei, China
Ischemia/reperfusion injury (IRI) is an inflammatory response that occurs when tissue is reperfused following a prolonged period of ischemia. Several studies have indicated that C-reactive protein (CRP) might play an important role in inducing IRI. However, the effects of CRP on myocardial IRI and the underlying mechanisms have not been fully elucidated. This study aimed to investigate the association between CRP and myocardial IRI and the underlying mechanisms.We simulated ischemia/reperfusion using oxygen-glucose deprivation/ reoxygenation (OGD/R) in neonatal Sprague-Dawley rat cardiomyocytes; reperfusion injury was induced by three hours of hypoxia with glucose and serum deprivation followed by one hour of reperfusion. Cell viability was tested with MTS assays, and cardiomyocyte damage was evaluated by lactate dehydrogenase (LDH) leakage. Mitochondrial membrane potential was measured using tetramethylrhodamine ethyl ester (TMRE) and mitochondrial permeability transition pore (mPTP) opening was measured using calcein/AM; both TMRE and caocein/AM were visualized with laser scanning confocal microscopy. In addition, we studied the signaling pathways underlying CRP-mediated ischemia/reperfusion injury via Western blot analysis.Compared with the simple OGD/R group, after intervention with 10 μg/mL CRP, cell viability decreased markedly (82.36 % ± 6.18%64.84% ± 4.06%,= 0.0007), and the LDH leakage significantly increased (145.3 U/L ± 16.06 U/L208.2 U/L ± 19.23 U/L,= 0.0122). CRP also activated mPTP opening and reduced mitochondrial membrane potential during myocardial ischemia/reperfusion. Pretreatment with 1 μM atorvastatin (Ator) before CRP intervention protected cardiomyocytes from IRI. Mitochondrial KATPchannel opener diazoxide and mPTP inhibitor cyclosporin A also offset the effects of CRP in this process. The level of phosphorylated extracellular-signal-regulated kinase (ERK) 1/2 was significantly higher after pre-treatment with CRP compared with the OGD/R group (170.4% ± 3.00%. 93.53% ± 1.94%,< 0.0001). Western blot analysis revealed that Akt expression was markedly activated (184.2% ± 6.96%. 122.7% ± 5.30%,= 0.0003) and ERK 1/2 phosphorylation significantly reduced after co-treatment with Ator and CRP compared with the level after CRP pretreatment alone.Our results suggested that CRP directly aggravates myocardial IRI in myocardial cells and that this effect is primarily mediated by inhibiting mitochondrial ATP- sensitive potassium (mitoKATP)channels and promoting mPTP opening. Ator counteracts these effects and can reduce CRP-induced IRI. One of the mechanisms of CRP-induced IRI may be related to the sustained activation of the ERK signaling pathway.
J Geriatr Cardiol 2018; 15: 492?503. doi:10.11909/j.issn.1671-5411.2018.07.001
C-reactive protein; Ischemia/reperfusion injury; Mitochondrial permeability transition pore; Mitochondrial KATPchannel; Statin
1 Introduction
In recent years, with the wide application of thrombolytic therapy and primary percutaneous coronary intervention (PCI), reperfusion therapies have become an effective treatment for ischemic heart disease. The mechanisms leading to the pathogenesis of myocardial ischemia/reperfusion injury (IRI) are complex and multifactorial, including pH lowering, marked oxidative/nitrosative stress, mitochondrial dysfunction, intracellular calcium overload and neurohormonal stimuli.[1]In most instances, reperfusion therapies can protect patients from myocardial necrosis and other related complications. Despite this improvement, sometimes reperfusion can also lead to the destruction of cardiac structure or function, which is generally referred to as myocardial IRI. The clinical manifestations include malignant arrhythmias, sudden drops in blood pressure, cardiac insufficiency and even sudden death, which seriously threaten to patients’ health, and we still do not have a definitive intervention to eliminate reperfusion-induced myocardial damage.
Inflammation is one of the most vital mechanisms when myocardial ischemia/reperfusion occurs. Oxidant induced leukocyte/endothelial cell interactions are largely responsible for the microvascular dysfunction induced by reperfusion, and proinflammatory neutrophils infiltrate ischemic tissues to exacerbate ischemic injury. C-reactive protein (CRP), an acute-phase plasma protein synthesized by the liver, commonly used as a marker for an acute inflammatory response, may be involved in myocardial ischemia/re-perfusion directly, due to its inflammatory property. There are few reports on the relationship between CRP and myocardial IRI.[2–4]Barrett,. demonstrated that the endogenous increase in plasma CRP secondary to a remote inflammatory lesion was associated with an increase in myocardial tissue injury secondary to regional ischemia and reperfusion. Dibra,.[3]indicated that CRP level on admission may predict the efficacy of reperfusion in patients with acute myocardial infarction and it was confined to patients treated with thrombolysis. However, whether CRP plays a direct role in aggravating IRI remains an open question, and the underlying mechanisms are not well understood.
The mitochondrial permeability transition pore (mPTP) and mitochondrial ATP- sensitive potassium (mitoKATP) channel are the two pivotal action sites in ischemia/re-perfusion injury. But the relationships between CRP and these two channels are still uncertain. In addition, a number of signaling pathways play pivotal roles in ischemia/re-perfusion. Nevertheless, information is scarce regarding the connection between CRP and these signaling pathways.
Statins have a wide range of biological effects in addition to reducing lipid levels. The Justification for Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) study placed emphasis on ostensibly healthy people with elevated high-sensitivity CRP (hsCRP) and decreased low-density lipoprotein (LDL) cholesterol levels who benefited from statin therapy.[5]The results showed that statin treatment was associated with a reduction of hsCRP and decreased incidence of cardiovascular events and all-cause mortality. However, little information is available regarding the relationship between statins and the process of CRP-mediated IRI.
We hypothesized that CRP, as a stimulating factor, could aggravate myocardial IRI. To test this hypothesis, we simulated ischemia/reperfusion using oxygen-glucose deprivation/ reoxygenation (OGD/R) in isolated rat cardiomyocyte primary cultures in the absence or presence of CRP and/or atorvastatin (Ator). We sought to investigate the mechanisms and signaling pathways involved in CRP-mediated IRI. Furthermore, we also aimed to find the possible signaling pathways involved in the protective effects of Ator against CRP-induced myocardial IRI.
2 Methods
2.1 Ethics statement
The protocol was approved by the Ethics Committee of Animal Experiments of Hebei medical university, and all animals received treatment in compliance with the Guide for the Care and Use of Laboratory Animals.
All steps in the study were performed in accordance with ethical standards. Every effort was made to reduce the number of animals and degree of pain in this experiment.
2.2 Drugs and reagents
Human recombinant CRP (Calbiochem, catalog number: 236608) was used. Collagenase (type II) was purchased from Gibco, Inc. (USA). Diazoxide (DZ), a selective mitoKATPchannel agonist, and cyclosporin A (CsA), an inhibitor of mPTP, both were obtained from Sigma Chemical Co. (St. Louis, MO). DZ and CsA were dissolved in dimethyl sulfoxide before being added into experimental solutions. The final concentration of dimethyl sulfoxide was < 0.1%. A CellTiter 96* nonradioactive cell proliferation assay (MTS assay) kit was purchased from Promega Corporation (Madison, WI). Lactate dehydrogenase (LDH) release was measured using a diagnostic kit (Jiancheng Bioengineering Institute, Nanjing, China). CRP we used was a pure pentamer, verified by native polyacrylamide gel electrophoresis (PAGE). To detect any bacterial contamination of CRP preparations, we removed endotoxin from the commercial CRP with a Limulus assay (XIAMEN BIOENDO Technology Co.) performed according to the manufacturer’s instructions. Briefly, the sample was adjusted for pH before purification. Then, the resin was activated and balanced. For endotoxin removal, after sample was added, the velocity of the flow controller was maintained at less than 0.25 mL/min. After 1.5 mL of liquid product accumulated, the sample was accepted by the non-heat receiver. The detection limit of this assay was 0.01 endotoxin units (EU)/mL. This purification was followed by plasma dilution and heat inactivation steps to diminish interference from plasma proteins. After purification, the endotoxin concentration in the CRP solution was < 0.1 EU/mL as measured by the Limulus assay.
2.3 Cell isolation and culture
Ventricular cardiomyocytes from neonatal Sprague-Daw-ley rats [postnatal day (PND) 1 to 2] were prepared. Pups were disinfected with 75% alcohol and the hearts were removed aseptically. The ventricles from eight to ten hearts from Sprague-Dawley rats were combined and the cardiac myocytes were isolated by trypsin and collagenase (type II) digestion, and cultured in high glucose Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) with 10% fetal bovine serum (FBS) (PAN, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin (Gibco, USA). The cells were plated at a density of 105cells/mL on 96-well plates in a humidified incubator (37 °C with 5% CO2) for 72 hours and the medium was changed after 48 hours. 5-Bromo-2’-deo-xyuridine (Brdu, Sigma, 0.1 mM) was added to the medium to inhibit fibroblast attachment and proliferation. As examined by immunofluorescence with antibodies against α- sarcomeric actin and α-smooth muscle actin, > 95% of the isolated cells were cardiomyocytes.
2.4 Oxygen-glucose deprivation and reoxygenation
Cell cultures were subjected to three hours OGD followed by one hour of reoxygenation. To simulate ischemia, glucose-free DMEM medium (Gibco, USA) was pre- equilibrated in 95% N2and 5% CO2at 37 °C. Oxygenated medium was removed from the cardiac myocyte cultures, and the N2-preequilibrated glucose-free DMEM was prom-ptly added. Cultures were immediately placed in a hypoxia container and exposed to 95% N2and 5% CO2for three hours at 37 °C. For reoxygenation, the glucose-free DMEM was replaced with high-glucose DMEM, and the cardio-myo-cy-tes were cultivated in 21% O2–5% CO2–74% room air for 1 hour at 37 °C. Cardiac myocytes unexposed to OGD/R served as the normoxic control.
2.5 Determination of CRP concentration
The CRP concentrations used in previous studies were commonly 5 μg/mL to 50 μg/mL. Therefore, we tested different final concentration of CRP starting at 5 μg/mL. Our pre-experiments showed that incubating cardiac myocytes with a final concentration of 10 μg/mL CRP before OGD has significant effects on ischemia/reperfusion injury, as established by MTS assays ( Figure 1A).
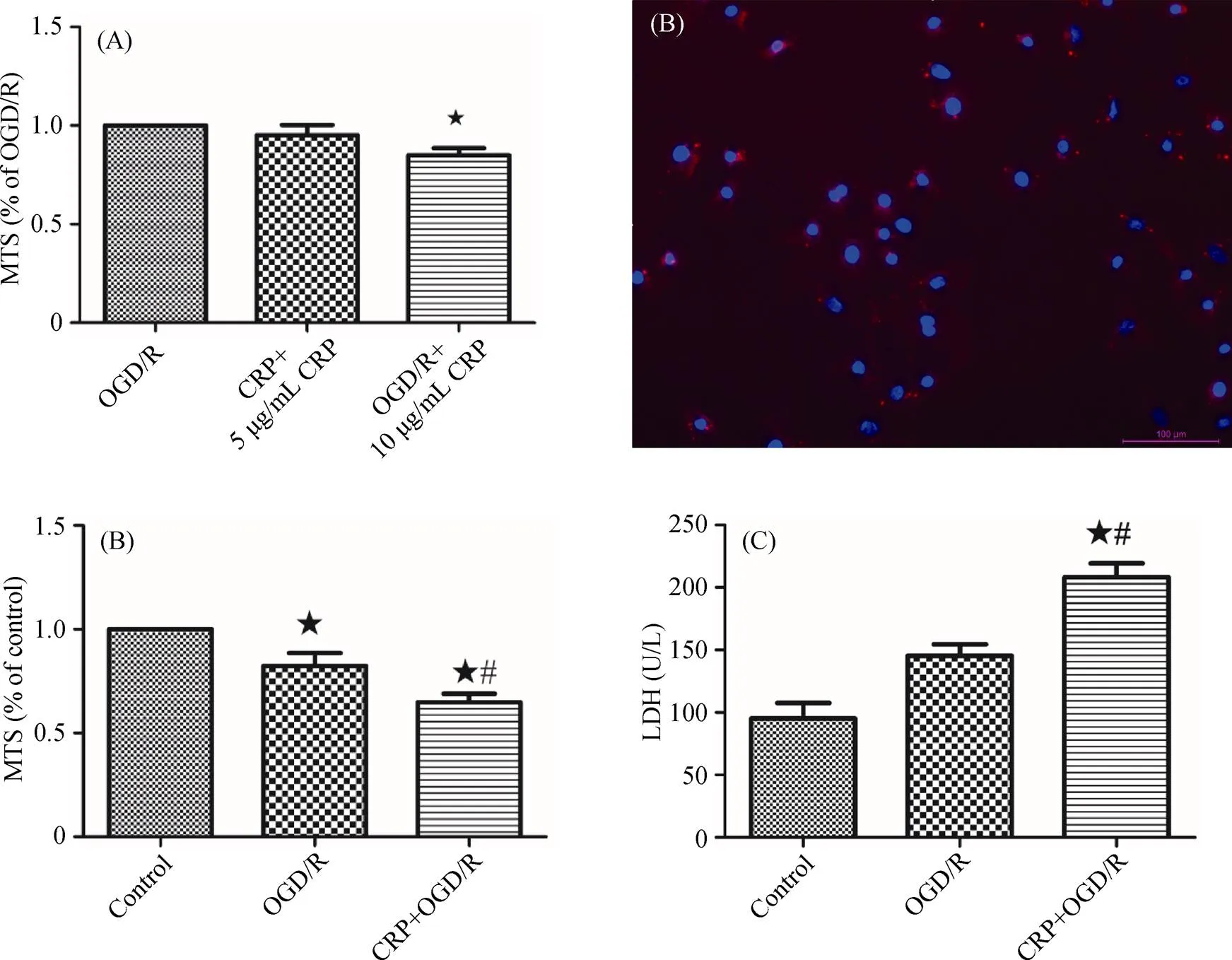
Figure 1. CRP aggravated myocardial IRI. (A): The cardiomyocytes incubated with 10 μg/mL of CRP before OGD have a significant effect on IRI; (B): immunofluorescence staining of neonatal rat cardiomyocytes with anti-α-actin antibody (red) and DAPI for nuclei (blue); (C): analysis of the effect of CRP on cell viability, detected by MTS assay; (D): effect of CRP on LDH levels in primary cardiomyocytes after OGD/R. Data were presented as mean ± SD.★< 0.05 compared with the control group;#< 0.05 compared with OGD/R group. CRP: C-reactive protein; IRI: ischemia reperfusion injury; LDH: lactate dehydrogenase; MTS: 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymeth-oxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; OGD: oxygen-glucose deprivation; OGD/R: oxygen-glucose deprivation/reoxygenation.
2.6 Experimental protocols
Human recombinant CRP was used in all studies described above. In addition, our pre-experiments have shown that Ator (1 μM) addition to cultured myocardial cells for three hours before OGD exerts the best cardioprotective effects. DZ selectively opens mitoKATPchannels but has no effect on cardiac sarcolemmal KATPchannels. In contrast, cyclosporin A is an inhibitor of mPTP.
Primary cultured neonatal rat myocardial cells were ran-domly divided into the following 6 groups: (1) control group: cardiomyocytes were cultured under normal conditions in high-glucose DMEM, in room air at 37 °C; (2) OGD/R group: after 72 hours of culture in high-glucose DMEM, cell samples were subjected to 3 hours OGD and 1 hour of reoxygenation; (3) CRP + OGD/R group: before OGD/R, cardiomyocytes were incubated with 10 μg/mL CRP for 24 hours; (4) Ator + CRP + OGD/R group: cells were pre-con-ditioned with 1 μM Ator for three hours, 10 μg/mL CRP for 24 hours, and then subjected to OGD/R; (5) CRP + CsA + OGD/R group: cardiomyocytes were treated with 10 μg/mL CRP for 24 hours and then subjected to three hours OGD. After the onset of reoxygenation, the cells were exposed to 10 μM CsA for 20 minutes; (6) CRP + DZ + OGD/R group: cells were incubated with 10 μg/mL CRP for 24 hours, washed with medium to remove CRP, and then exposed to 100 μM diazoxide for 90 minutes before OGD/R.
2.7 Cardiomyocyte identification
After being cultured for 72 hours, the cell samples were rinsed with polybutylene succinate (PBS), fixed with 4% paraformaldehyde for 20 minutes, and then permeabilized with glycine/triton solution (PBS/0.5% Triton/10 mM glycine). The samples were incubated with mouse monoclonal anti-α-actin (1:300 dilution) (Sigma; catalog number: A7811) at 4 °C overnight. After rinses, the samples were incubated with anti-mouse IgG- FITC (1:300 dilution) (Sigma; catalog number: F2012) for 60 minutes at 37 °C. Nuclei were counter-stained with 100 nM diamidine pheny-lindole (DAPI) (Invitrogen) for 30 minutes followed by rinses with PBS. Fluorescent images were observed with a spectral confocal microscope imaging system (Leica TCS SP2).
2.8 Quantification of cell biability by MTS assays
Cell viability was measured with the MTS assay. After OGD/R treatment, 20 μL of MTS solution (5 mg/mL) was added to each well, and the cardiomyocytes were maintained in the growth medium for 1 hour at 37 °C. Absorbance was measured at 490 nM using a microplate reader (M200PRO, TECAN, Austria). The cells without treatment were considered controls. Moreover, the growth medium without cardiomyocytes in the presence of MTS solution was used as a solution background. Cell viability was calculated as the percentage of the control group, depending on different situations. There were five samples in each group, and the experiment was repeated at least three times.
2.9 LDH leakage assay
As a marker for myocardial necrosis, LDH release was measured in the culture medium using a diagnostic kit. The total LDH activity was determined after freezing each culture at-80 °C overnight and subsequently thawing the cultures rapidly when needed. The experiment was repeated at least three times.
2.10 Observation of mitochondrial membrane potential (ΔΨm) by laser scanning confocal microscopy
The fluorescence intensity of tetramethylrhodamine ethyl ester (TMRE, Invitrogen) of the isolated cardiomyocytes was determined to estimate the value of mitochondrial membrane potential caused by hypoxia and reoxygenation. TMRE is a cationic dye that accumulates in polarized mitochondria and is released if mitochondria depolarize. To measure mitochondrial membrane potential, the cardiomyocytes were washed with PBS and loaded with 100 nM TMRE for 20 minutes at room temperature away from light. The fluorescence intensity of TMRE was measured using an LSM-510 laser scanning confocal microscope (Leica, Germany) with 543 nM excitation and 580 nM emission. All images were analyzed using LSM-510 V.2.3 software. Fluorescent intensities were expressed as percent changes compared with the control group value.
2.11 Measurement of mitochondrial mPTP opening by laser scanning confocal microscopy
In addition to TMRE, calcein, a membrane potential-in-de-pendent tracer, was used to evaluate the opening of the mPTP, which remains closed during myocardial ischemia and opens in the first few minutes of reperfusion. Therefore, we chose to measure fluorescence intensity in fluorescent probe-loaded cardiomyocytes 20 minutes after reoxygenation. After reoxygenation for 20 minutes, cells were loaded with calcein/AM (1.0 μM) and cobalt chloride (CoCl2) (2.0 mM) for 20 minutes at room temperature and then washed with PBS. The calcein/AM is distributed into the mitochondria and cytosol, but the cytosolic calcein is quenched by CoCl2, leaving only the calcein fluorescence in mitochondria visi-ble. Images of calcein fluorescence (excitation at 488 nM and emission at 505 nM) were acquired using a laser scanning confocal microscope (LeicaTCSSP5, Mannheim, Germany).
2.12 Western blot analysis
Cytoplasmic proteins were prepared from heart tissues, and immunoblots were performed as previously described. In brief, tissue proteins were obtained from the heart in the first four groups and were lysed in ice-cold extraction buffer containing a protease inhibitor cocktail for 30 min. The whole lysates were then centrifuged at 8000 × g for 10 min, and the protein concentration in the supernatant was determined using a modified Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA). Protein samples were separated by electrophoresis via sodium dodecyl sulfate (SDS)-PAGE, transferred to polyvinylidene fluoride (PVDF) membranes (Beyotime, China), and incubated overnight at room temperature in tris-buffered saline (TBS) with 5% nonfat milk. After blocking, the membranes were probed with primary antibodies (1:1000) against Akt (Abcam, California, USA), extracellular signal-regulated kinase 1/2 (ERK 1/2, Cell Signaling Technology, California, USA), phosphorylated ERK 1/2 (T202/ Y204, Santa Cruz, CA, USA), p38 mitogen activated pro-tein kinase (MAPK), c-Jun N-terminal kinase (JNK) (Cell Signaling Technology, California, USA), and β-actin (Santa Cruz, CA, USA) and then incubated with corresponding horseradish peroxidase-conjugated secondary antibodies (Sigma-Aldrich) for one hour at room temperature. Chemi-lu-minescence was induced by the addition of the SuperSig-nal West Pico Chemiluminescent Substrate (Pierce, Rock-ford, IL) and observed using radiographic film. Band inten-sity was quantified using ImageJ 1.50 software. The levels of Akt, ERK 1/2, p38 MAPK and JNK were normalized to that of β-actin.
2.13 Statistical analysis
Continuous variables were reported as the mean ± SD if normally distributed. Comparisons between two groups were evaluated using student’s-test. One-way ANOVA were used for categorical variables. One-way ANOVA was used to assess differences between groups. In all cases, statistical significance was set at< 0.05. Statistical analysis was performed with GraphPad Prism (version 4.0, GraphPad Software Inc).
3 Results
3.1 CRP aggravated myocardial IRI
To evaluate the effects of CRP on myocardial IRI, we first validated neonatal rat cardiac ventricular cardiomyocytes (Figure 1B). Then, the cardiomyocytes were pretreated with 10 μg/mL CRP for 24 hours, and then subjected to three hours of OGD, and provided one hour of reoxygenation. The cell viability of cardiomyocytes was determined by the MTS assay. As shown in Figure 1C, OGD/R-induced damage significantly decreased cell viability to 82.36 % ± 6.18% of the control (= 0.0031), which indicated that our experimental protocols of three hours OGD and 1hour reoxygenation successfully stimulates myocardial IRI in vitro. Intervention with 10 μg/mL CRP further decreased the cell viability to 64.84% ± 4.06% of the control (< 0.0001), and the cell viability in the CRP + OGD/R group was significantly different from the value in the OGD/R group (= 0.0007). Similarly, a significant difference was observed in LDH leakage in the CRP pretreatment group compared with the OGD/R group lacking CRP (208.2 U/L ± 19.23 U/L versus 145.3 U/L ± 16.06 U/L,= 0.0122), as depicted in Figure 1D. These data suggested that CRP treatment had a significant effect on myocardial ischemia/reperfusion. CRP can aggravate IRI.
3.2 CRP reduced the mitochondrial membrane potential and promoted mPTP opening in myocardial ischemia/reperfusion
It is well established that mPTP opening is responsible for the pathogenesis of ischemia/reperfusion and that mitoKATPopening is essential for cardioprotection; the decline of mitochondrial membrane potential is one of the hallmarks of apoptosis. Thus, to clarify the relationships among CRP, mPTP opening, and mitochondrial membrane potential, we measured the changes of calcein/AM and TMRE fluorescence in control, OGD/R, and CRP + OGD/R groups by laser scanning confocal microscopy. These changes can be directly observed in Figure 2A and Figure 3A. After treatment with 10 μg/mL CRP, the average fluorescence intensity values of calcein/AM significantly decreased compared with the OGD/R groups (33.08% ± 3.48%. 60.93% ± 2.82%,< 0.0001) and the TMRE group (33.31% ± 2.03%. 57.26% ± 2.95%,< 0.0001), as shown in Figure 2B and Figure 3B, which indicated that CRP activated mPTP opening and reduced mitochondrial membrane potential during ischemia/reperfusion.
3.3 Atorvastatin conferred protection against CRP- me-diated myocardial IRI
To further understand and validate the effect of Ator in CRP-mediated myocardial IRI, cultured cardiomyocytes were incubated with 1 μM Ator before exposure to 10μg/mL CRP, and subsequent OGD/R. After OGD/R, the cell viabil-ity of cardiomyocytes was assessed with MTS assays. Ator significantly increased cell viability, compared with the CRP + OGD/R group (98.61% ± 5.01%. 64.84% ± 4.06% of control viability;< 0.0001). Similarly, Ator signifi-cantly decreased LDH leakage compared with pretreatment with CRP alone before OGD/R (94.35 U/L ± 14.83 U/L. 208.20 U/L ± 19.23 U/L,= 0.0013). In addition, we used a specific mitoKATPopener, DZ, at 100 μM, and a specific mPTP inhibitor, CsA, at 10 μM to reverse the effects of CRP in ischemia/reperfusion. The results mentioned above are shown in Figure 4A and Figure 4B. Laser scanning confocal microscopy analysis was also performed to quantify changes in the levels of mPTP opening and mitochondrial membrane potential in response to Ator treatment. As mentioned previously, CRP significantly reduced the fluorescence intensity of calcein/AM. However, pretreatment with 1 μM Ator for three hours significantly increased fluorescence intensity compared with the CRP + OGD/R group (92.74% ± 2.47%. 33.08% ± 3.48%,< 0 .0001), as shown in Figure 5A and Figure 5B, indicating that Ator inhibited mPTP opening in cardiomyocytes. Consistent with the results of calcein/AM, co-treatment with Ator and CRP increased TMRE fluorescence intensity observably compared with simple treatment with CRP before OGD/R (84.00% ± 2.31%. 33.31% ± 2.03%,< 0.0001), as depicted in Figure 6A and Figure 6B. At the same time, the CRP-mediated decrease in calcein/AM and TMRE fluorescence was completely offset by exposure to CsA or DZ. These results suggested that CRP aggravated IRI by inhibiting mitoKATPand further promoting mPTP opening. We also verified that Ator protected cardiomyocytes from CRP-mediated IRI by reducing the loss of mitochondrial membrane potential and inhibiting mPTP opening.

Figure 2. CRP promoted the opening of mPTP. (A): effect of CRP on the calcein fluorescence by confocal laser scanning microscope. Compared with control group, OGD/R significantly reduced the intensity of fluorescence. The pretreatment with CRP (10 μg/mL) for 24 hours further decreased fluorescence intensity significantly. (B): the specific values of calcein/AM fluorescent intensities in above groups were shown. Data were presented as mean ± SD.★< 0.05 compared with control group;#< 0.05 compared with OGD/R group. AM: aminomethyl; CRP: C-reactive protein; mPTP: mitochondrial permeability transition pores; OGD/R: oxygen-glucose deprivation/reoxygenation.
Figure 3. CRP aggravated depolarization of mitochondrial membrane potential in cardiomyocytes. (A): effect of CRP on the TMRE fluorescence by confocal laser scanning microscope. Compared with control group, OGD/R significantly reduced the intensity of fluorescence. The pretreatment with CRP (10 μg/mL) for 24 hours further decreased fluorescence intensity significantly. (B): the specific values of TMRE fluorescent intensities in above groups were shown. Data were presented as mean ± SD.★< 0.05 compared with control group;#< 0.05 compared with OGD/R group. CRP: C-reactive protein; OGD/R: oxygen-glucose deprivation/ reoxygenation; TMRE: tetramethylrhodamine ethyl ester.
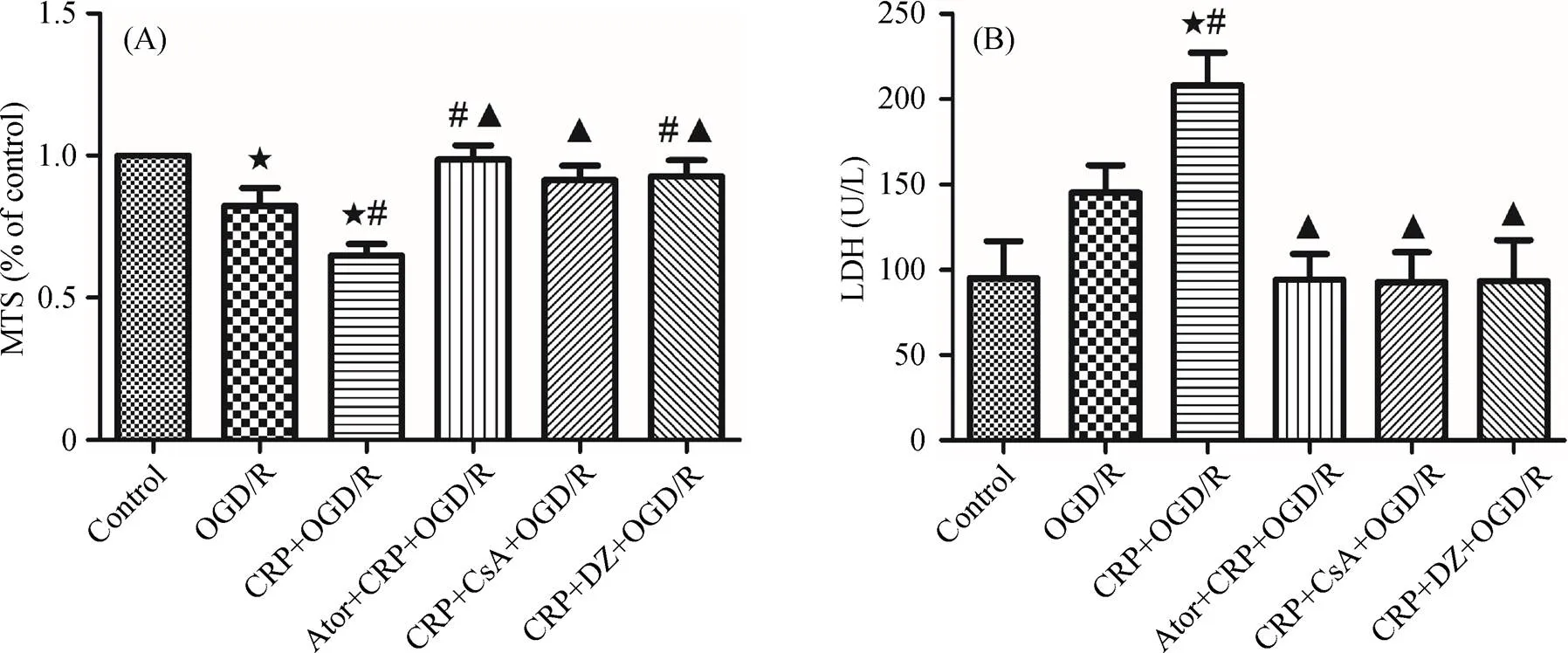
Figure 4. Ator protected cardiomyocytes from CRP-mediated IRI. (A): analysis of the effects of Ator, CsA and DZ on cell viability, detected by MTS assay. SD rat cardiomyocytes were incubated with CRP at concentration of 10 μg/mL for 24 hours, aggravating cell injury induced by OGD/R. This adverse effect of CRP was blocked by CsA (10 μM) or DZ (100 μM). Ator (1 μM) preconditioned primary cardiomyocytes against CRP-induced cell injury. (B); effects of Ator, CsA and DZ on LDH level in primary cardiomyocytes after OGD/R. Data were presented as mean ± SD.★< 0.05 compared with control group;#< 0.05 compared with OGD/R group;▲< 0.05 compared with CRP+OGD/R group. Ator: Atorvastatin; CRP: C-reactive protein; CsA: cyclosporin A; DZ: diazoxide; IRI: ischemia reperfusion injury; LDH: lactate dehydrogenase; OGD/R: oxygen-glucose deprivation/reoxygenation.
Figure 5. Ator alleviated mPTP opening in cardiomyocytes. (A): effect of Ator on the calcein fluorescence by confocal laser scanning microscope. Pretreatment with Ator (1 μM) for three hours resulted in a significant increase in fluorescence intensity. In addition, CsA (10 μM) or DZ (100 μM) inhibited the CRP-induced decrease of fluorescence intensity. (B): the specific values of calcein/AM fluorescent intensities in above groups were shown. Data were presented as mean ± SD.★< 0.05 compared with control group;#< 0.05 compared with OGD/R group;▲< 0.05 compared with CRP + OGD/R group. AM:aminomethyl; Ator: Atorvastatin; CRP: C-reactive protein; CsA: cyclosporin A; DZ: diazoxide; mPTP: permeability transition pores; OGD/R: oxygen-glucose deprivation/reoxygenation.
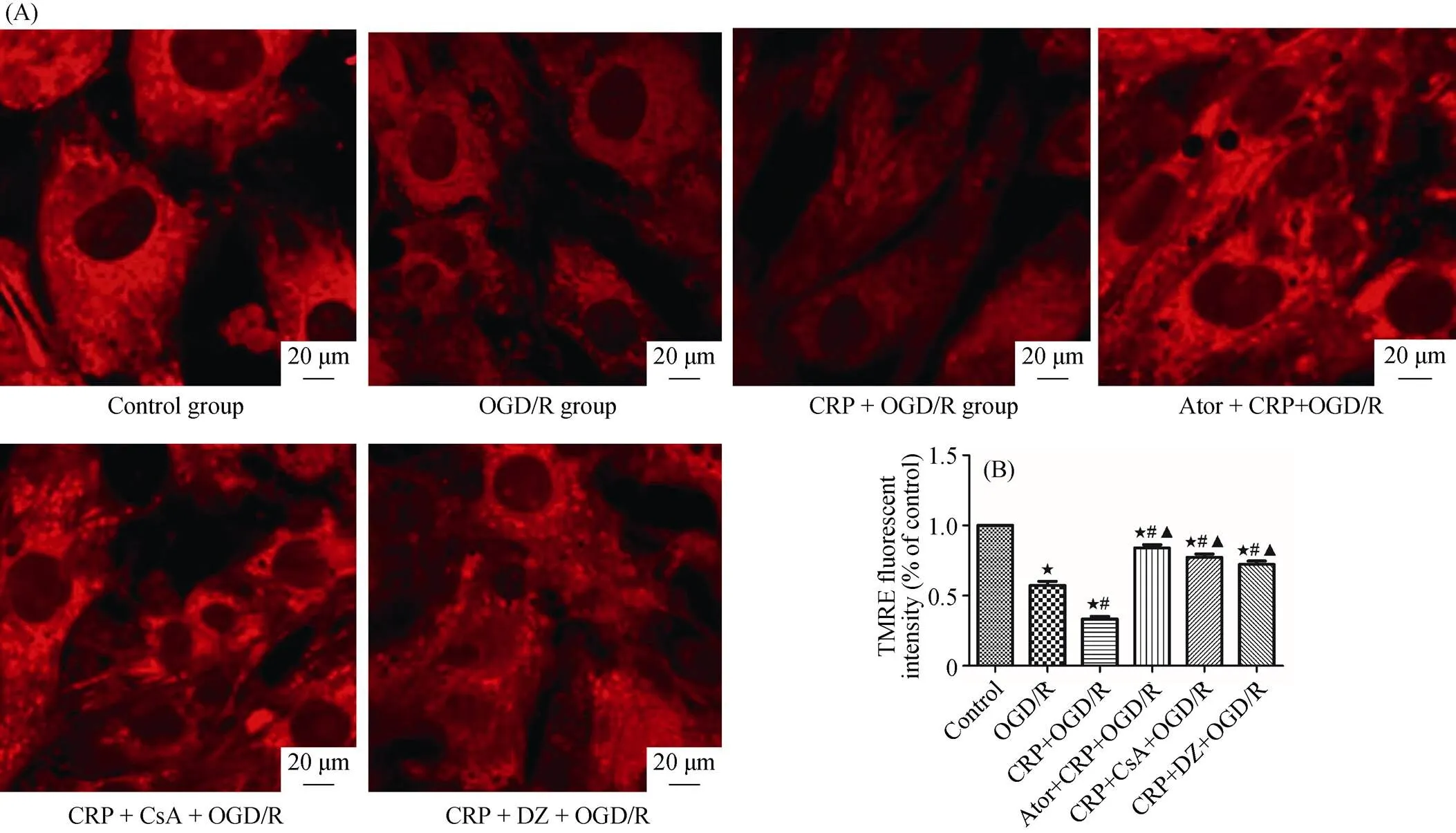
Figure 6. Ator prevented depolarization of mitochondrial membrane potential in cardiomyocytes. (A): effect of Ator on the TMRE fluorescence by confocal laser scanning microscope. Compared with CRP + OGD/R group, the pretreatment with Ator (1 μM) for three hours resulted in a significant increase in fluorescence intensity. In addition, cotreatment with CsA (10 μM) or DZ (100 μM) inhibited the CRP-induced decrease of fluorescence intensity. (B): the specific values of calcein/AM fluorescent intensities in above groups were shown. Data were presented as mean ± SD.★< 0.05 compared with control group;#< 0.05 compared with OGD/R group;▲< 0.05 compared with CRP + OGD/R group. AM:aminomethyl; Ator: Atorvastatin; CRP: C-reactive protein; CsA: cyclosporin A; DZ: diazoxide; OGD/R: oxygen-glucose deprivation/ reoxygenation; TMRE: tetramethylrhodamine ethyl ester.
3.4 CRP aggravated IRI primarily through ERK signaling pathway activation, and atorvastatin protected cardiomyocytes via the Akt and ERK signaling pathways
To further explore the signaling pathways that mediate the effects of CRP on myocardial ischemia/reperfusion, we examined the expression of ERK 1/2, Akt, JNK and p38 MAPK after CRP treatment by Western blot analysis. As shown in Figure 7, the levels of phosphorylated ERK 1/2 (phospho-ERK 1/2) and total ERK 1/2 were both markedly increased after treatment with CRP. The difference was especially significant for phospho-ERK 1/2 (93.53% ± 1.94%. 170.4% ± 3.00%,< 0.0001). However, no remarkable changes of the total levels of p38 MAPK and JNK were observed after CRP treatment followed by OGD/R compared with the OGD/R group. These results suggested that the upregulation of phospho-ERK and total ERK are involved in CRP-mediated ischemia/reperfusion injury in cardiomyocytes. In our experiment, we found that Ator attenuated CRP-mediated ischemia/reperfusion injury in cardiac myocytes, but the specific mechanisms remained unclear. Accordingly, one purpose of this part of the study was to explore the possible signaling pathways that mediated the effect of Ator on CRP-induced IRI. Western blot analysis revealed that Akt expression was markedly activated after co-treatment with Ator and CRP compared with the level after simple CRP pretreatment (184.2% ± 6.96%. 122.7% ± 5.30%,= 0.0003). In addition, the levels of both ERK and phospho-ERK were lower in the Ator and CRP co-treatment group compared with the levels in CRP group after OGD/R. The difference was especially significant for phospho-ERK. In summary, we concluded that Ator protected cardiomyocytes from CRP-mediated ischemia/re-perfusion injury mainly by activating the Akt signaling pathway and decreasing phosphorylated ERK levels.
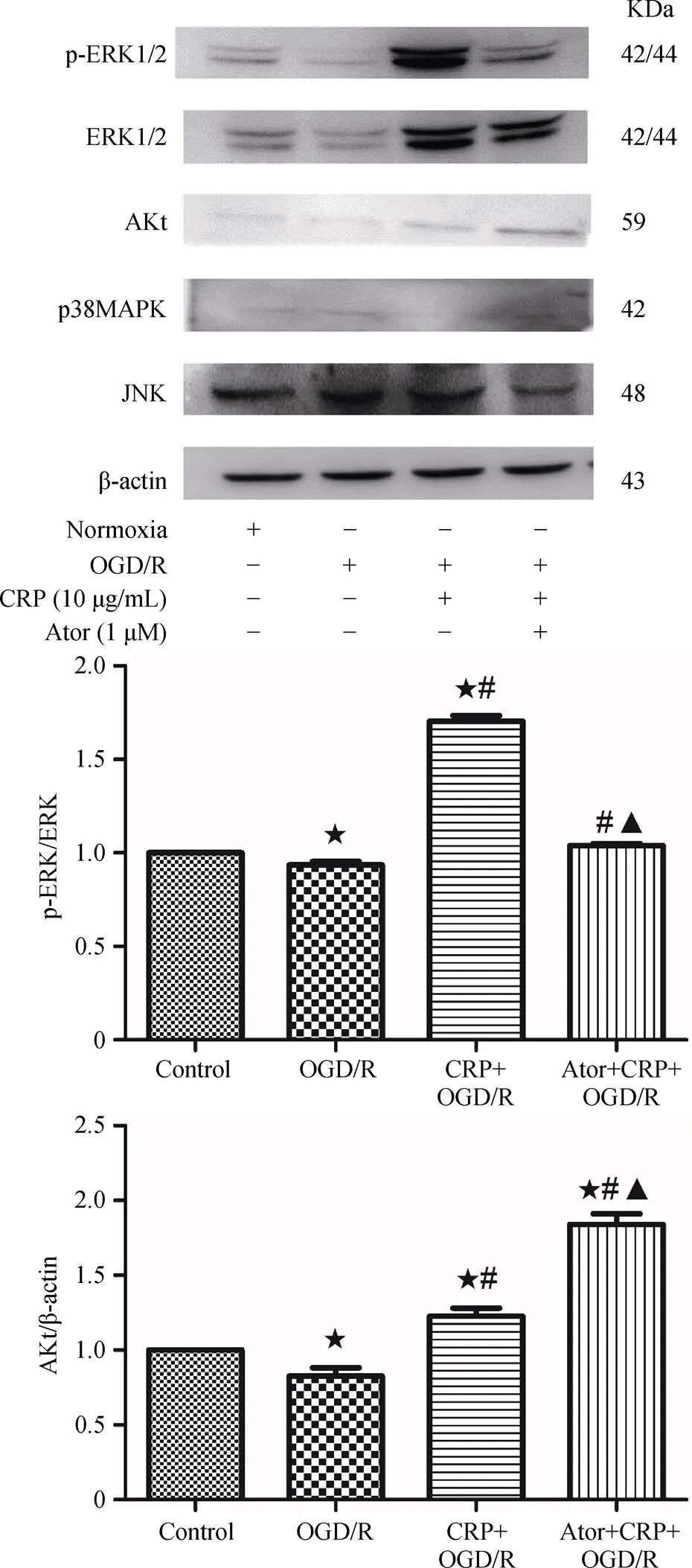
Figure 7. Analysis of the effects of CRP and Ator on related protein expressions, detected by western blot. β-actin was used as a loading control. These results indicated that CRP aggravated myocardial ischemia reperfusion mainly via activating the ERK signaling pathway, and Atorvastatin protected cardiomyocytes from CRP-induced ischemia reperfusion through the Akt and ERK Signaling Pathway.★< 0.05 compared with control group;#< 0.05 compared with OGD/R group;▲< 0.05 compared with CRP + OGD/R group. Ator: Atorvastatin; CRP: C-reactive protein; ERK: extracellular-signal-regulated kinase; JNK: Jun N-terminal kinase; MAPK: mitogen activated protein kinase; OGD/R: oxygen-glucose deprivation/ reoxygenation; p-ERK.
4 Discussion
The main findings of our present study are: (1) CRP di-rectly aggravated myocardial ischemia/reperfusion by in-hibiting the mitoKATPchannel and mPTP opening; (2) there was a significant increase in the extracellular sig-nal-re-gulated kinase 1/2 (ERK 1/2) and phosphorylated ERK 1/2 (phospho-ERK 1/2) levels when cardiomyocytes were pre-treated with CRP before OGD/R. The effects of CRP in the process of ischemia/reperfusion may be related to the acti-vation of the ERK signaling pathway; and (3) atorvastatin protected cardiomyocytes from CRP-induced ischemia/re-perfusion injury mainly by activating the phosphatidylino-sitol-3-kinase (PI3K)/Akt/glycogen synthase kinase 3β (GSK-3β) signaling pathway and inhibiting the phospho- ERK signaling pathway.
Previous studies on the relationship between CRP and ischemia/reperfusion have mostly focused on the effects of CRP on renal ischemia/reperfusion.[6–8]Thiele,. showed that CRP was a potent modulator of renal IRI. Conformational changes of CRP accelerated leukocyte recruitment and reactive oxygen species (ROS) formation, accordingly aggravating renal IRI. It has also been reported that CRP-induced renal reperfusion injury was associated with down-regulating autophagy flux and shifting the balance of macrophage activation and FcγR expression. As for myocardial IRI, Thiele,.[9]suggested that CRP dissociation modulated inflammation in acute (cardiac ischemia/re-perfusion) and chronic (atherosclerosis) inflammatory processes. Barrett,.[2]observed the relationship between CRP treatment concentration and myocardial infarct size in rabbits, indicating that CRP aggravated myocardial IRI due to the activation of the complement cascade. Valtchanova-Matchouganska,.[10]confirmed the involvement of CRP as a marker in cardiac ischemic reperfusion injury, because the increase of CRP was prevented by both ischemic preconditioning and pharmacological (chemical) preconditioning in early, and especially in late preconditioning. Consistent with previous studies, we demonstrated that CRP directly aggravated myocardial IRI, and CRP intervention for 24 hours prior to oxygen-glucose deprivation/reoxygenation markedly decreased cell viability and increased LDH leakage into cardiac cell-culture medium. These results indicated that CRP directly exacerbated myocardial IRI, which provides a new therapeutic target to reduce reperfusion injury in clinical practice.
CRP has multiple biological activities including binding to specific receptors on leukocytes to modulate their function, the induction of the expression of adhesion molecules on endothelial cells, the activation of the classical complement pathway by binding to complement factor 1q (C1q), and calcium-dependent binding to phosphocholine, fibro-nectin, chromatin, histones, and ribonucleoprotein.[2]
Both the inhibition of mitoKATPand the opening of mPTP are closely associated with myocardial IRI. MPTP is quiescent during ischemia. Nevertheless, after reperfusion, the enormous increases in mitochondrial Ca2+with the addi-tion of an ROS burst, induces mPTP opening and accordingly produces a chain of events that results in cardiomyo-cyte death. The opening of mitoKATPchannels during is-chemia causes mitochondrial membrane depolarization and decreases mitochondrial membrane potential, thus resulting in protein kinase C (PKC) activation, the inhibition of ROS production, and consequent antiapoptotic effects.[11]DZ, a selective mitoKATPchannel opener, and CsA, an inhibitor of mPTP, have been confirmed to reduce myocardial IRI. In our study, we found that co-treatment of CRP with DZ or CsA before OGD/R offset the influences of CRP on cell viability and LDH leakage. In addition, mitochondrial membrane depolarization was implicated in cardiac myocyte death in the process of ischemia/reperfusion; thus, we measured ΔΨm using the indicator TMRE. Our study demonstrated that the reduced fluorescence intensity of TMRE in the CRP treatment group was significantly increased by the addition of DZ or CsA to the medium. Moreover, CRP directly promoted mPTP opening based on fluorescence intensity comparison, using the probe calcein/AM and CoCl2. Additional intervention with DZ or CsA blocked the promotion of mPTP opening by CRP. Taken together, these results suggested that both the mitoKATPchannel and the mPTP are involved in the mechanisms of CRP-mediated increased myocardial IRI. We confirmed our finding that CRP aggravated myocardial IRI and showed for the first time that these effects are likely to be mediated primarily via the inhibition of mitoKATPchannels and the promotion of mPTP opening.
Furthermore, in the current study, we found that activation of the ERK 1/2 signaling pathway mediated CRP-in-duced myocardial IRI in SD rat cardiomyocytes. Several studies have shown that CRP induces ERK activation in adipose-derived stem cells and human coronary artery endothelial cells.[12,13]Zhong,. reported that CRP stimulated the expression of the receptor for advanced glycation end-products in human coronary artery endothelial cells to mediate CRP-induced pro-atherosclerotic activation through ROS generation and the activation of the ERK signaling. Consistent with these previous results, we also found a significant increase in the ERK 1/2 and phosphorylated ERK 1/2 (phospho-ERK 1/2) levels when cardiomyocytes were pretreated with CRP for 24 hours before OGD/R, which indicated that the persistent activation of the ERK 1/2 signaling pathway was one of the possible mechanisms. Phospho-ERK 1/2 is an activated form. The increase in the level of phospho-ERK 1/2 after intervention with CRP was more significant than the increased expression of ERK 1/2 in our study. To explore whether JNK and p38 MAPK are closely related to CRP-induced myocardial ischemia/reperfusion, the expression levels of JNK and p38 MAPK after incubation with CRP were also detected. CRP treatment had no significant effects on JNK or p38 MAPK expression during ischemia/reperfusion. The possible mechanisms of CRP parti- cipating in myocardial ischemia/reperfusion are shown in Figure 8.
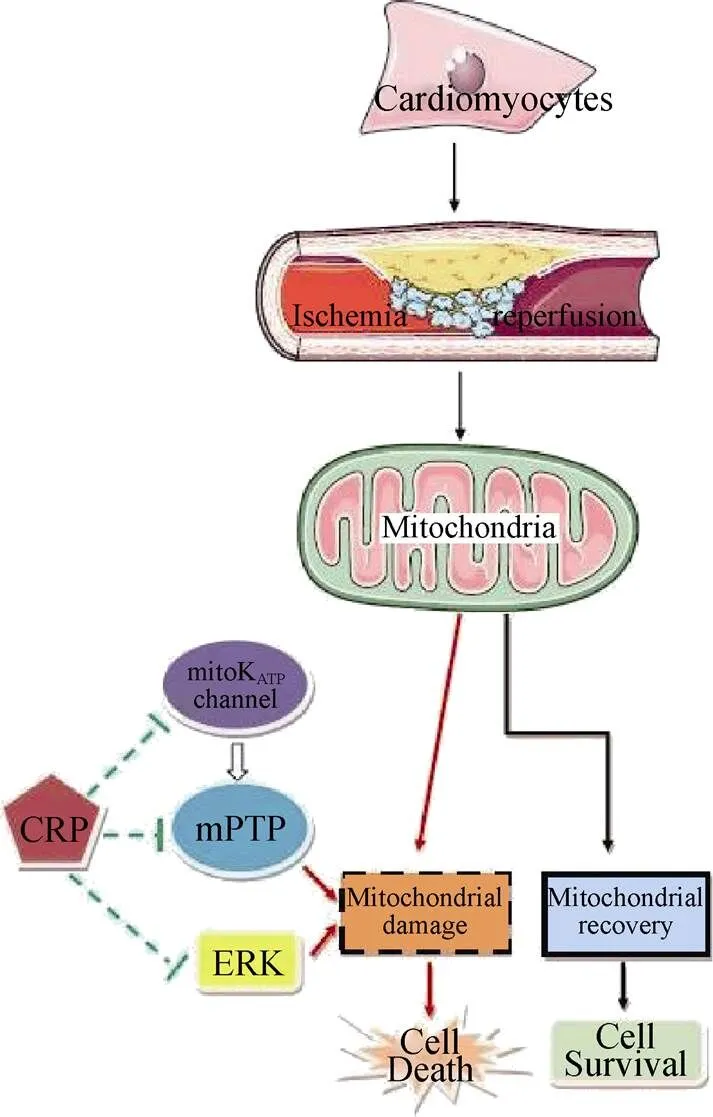
Figure 8. The schematic diagram of the related mechanisms involved in the CRP-induced ischemia reperfusion injury in cardiomyocytes. CRP: C-reactive protein; ERK: extracellular- sig-nal-regulated kinase; mitoKATP: mitochondrial ATP-sensitive po-tassium; mPTP: permeability transition pores.
In our study, we found that Ator decreased CRP-induced myocardial IRI, which may be largely related to its inhibition of the inflammatory response. We found that Ator prevented the mPTP opening and alleviated the collapse of the mitochondrial membrane potential in CRP-mediated IRI, which is consistent with our prior study on the effects of Ator on myocardial IRI in the absence of CRP.[14]In addition, we also found that pretreatment with Ator markedly activated the expression of Akt and reduced the level of phospho-ERK. The PI3K/Akt pathway has a protective effect against IRI. In addition, PI3K/Akt pathway activation decreases mitochondria-mediated apoptosis. Serine/threo-nine kinase Akt is a primary modulator of the downstream effects of PI3K, maintaining the integrity of the mitochondrial by phosphorylating molecules such as GSK-3β. Early research in our group has shown that Ator provides cardioprotective effects against IRI by increasing GSK-3β as a result of inhibiting miR-199a-5p.[15]Therefore, taken together, we can speculate that Ator reduced CRP-mediated myocardial IRI mainly by activating the PI3K/Akt pathway and then further increasing the expression of GSK-3β. In our experiment, the data indicated that pretreatment with Ator before CRP intervention and OGD/R decreased phospho-ERK expression significantly, which may be another mechanism involved in the signaling pathways in this process.
In conclusion, our study demonstrated that CRP aggravated myocardial IRI in primary cultures of SD rat myocardial cells and that this aggravation was mediated by the inhibition of mitoKATPchannels and the promotion mPTP opening. The increased IRI effects of CRP may be achieved by up-regulating the ERK signaling pathway. These findings may provide a new treatment for reducing IRI and provide additional benefit to patients with ischemic heart disease undergoing reperfusion therapies. Nevertheless, subsequent studies should be conducted to define the functions of CRP in myocardial ischemia/reperfusion and its effects in vivo, in addition to exploring other related signaling pathways involved in this process. Our findings also indicated that statins may play protective roles against CRP-induced myocardial IRI and provided evidence for the involvement of Akt activation and phospho-ERK inhibition in the atorvastatin-mediated alleviation of the pathology of IRI.
Acknowledgment
The study was supported partly bythe Health Department of Hebei Province Key Science and Technology Research program (20140073, Shijiazhuang, China) and the Science Research Foundation of the Second Hospital of Hebei Medical University (2h2201514, Shijiazhuang, China).
The authors declared no conflict of interests.
1 Sanada S, Komuro I, Kitakaze M,. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures.2011; 301: H1723–1741.
2 Barrett TD, Hennan JK, Marks RM,. C-reactive-pro-tein-associated increase in myocardial infarct size after ischemia/reperfusion.2002; 303: 1007–1013.
3 Dibra A, Mehilli J, Schwaiger M,. Predictive value of basal C-reactive protein levels for myocardial salvage in patients with acute myocardial infarction is dependent on the type of reperfusion treatment.2003; 24: 1128–1133.
4 Wang QQ, Qiu YG, Zhu YJ,. Cyclophosphamide protects against myocardial ischemia/reperfusion injury in rats: one of the therapeutic targets is high sensitivity C-reactive protein.2009; 137: 991–996.
5 Ridker PM, Danielson E, Fonseca FA,. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein.2008; 359: 2195–2207.
6 Thiele JR, Zeller J, Kiefer J,. A Conformational Change in C-Reactive Protein Enhances Leukocyte Recruitment and Reactive Oxygen Species Generation in Ischemia/Reperfusion Injury.2018; 9: 675.
7 Bian A, Shi M, Flores B,. Downregulation of autophagy is associated with severe ischemia-reperfusion-induced acute kidney injury in overexpressing C-reactive protein mice.2017; 12: e0181848.
8 Pegues MA, McCrory MA, Zarjou A,. C-reactive protein exacerbates renal ischemia-reperfusion injury.2013; 304: F1358–1365.
9 Thiele JR, Habersberger J, Braig D,. Dissociation of pentameric to monomeric C-reactive protein localizes and aggravates inflammation: in vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy.2014; 130: 35–50.
10 Valtchanova-Matchouganska A, Gondwe M, Nadar A,. The role of C-reactive protein in ischemia/reperfusion injury and preconditioning in a rat model of myocardial infarction.2004; 75: 901–910.
11 Wu H, Wang P, Li Y,. Diazoxide attenuates postresuscitation brain injury in a rat model of asphyxial cardiac arrest by opening mitochondrial ATP-sensitive potassium channels.2016; 2016: 1253842.
12 Zhong Y, Cheng CF, Luo YZ,. C-reactive protein stimulates RAGE expression in human coronary artery endothelial cells in vitro via ROS generation and ERK/NF-κB activation.2015; 36: 440–447.
13 Chen J, Gu Z, Wu M,. C-reactive protein can upregulate VEGF expression to promote ADSC-induced angiogenesis by activating HIF-1α via CD64/PI3k/Akt and MAPK/ERK signaling pathways.2016; 7: 114.
14 Zhao Z, Cui W, Zhang H,. Pre-treatment of a single high-dose of atorvastatin provided cardioprotection in different ischaemia/reperfusion models via activating mitochondrial KATP channel.2015; 751: 89–98.
15 Zuo Y, Wang Y, Hu H,. Atorvastatin protects myocar-dium against ischemia-reperfusion injury through inhibiting miR-199a-5p.2016; 39: 1021–1030.
Wei CUI, Department of Cardiology, Second Hospital of Hebei Medical University and Institute of Cardiocerebrovascular Disease of Hebei Province, Shijiazhuang, Hebei, China. E-mail: cuiwei2013@hotmail.com
Telephone: + 86-311-66002115
Fax:+ 86-311-66002115
May 26, 2018
June 16, 2018
June 19, 2018
June 28, 2018
 Journal of Geriatric Cardiology2018年7期
Journal of Geriatric Cardiology2018年7期
- Journal of Geriatric Cardiology的其它文章
- Single-territory incomplete surgical revascularization improves regional wall motion of remote ventricular areas: results from a propensity-matched study
- A three-year longitudinal study of the relation between left atrial diameter remodeling and atrial fibrillation ablation outcome
- Particular evolution in a 72-year-old diabetic patient with acute coronary syndrome
- Management of hypertensive crises in the elderly
- Prognostic utility of NT-proBNP greater than 70,000 pg/mL in patients with end stage renal disease
- The effectiveness and safety of the RESTORE? drug-eluting balloon versus a drug-eluting stent for small coronary vessel disease: study protocol for a multi-center, randomized, controlled trial