
Resistance to epidermal growth factor receptor inhibition in non-small cell lung cancer
2018-02-18 09:24:24NeleVanDerSteenElisaGiovannettiDanielaCarboneAlessandroLeonettiChristianRolfoGodefridusPeters
Cancer Drug Resistance 2018年4期
Nele Van Der Steen, Elisa Giovannetti, Daniela Carbone, Alessandro Leonetti,Christian D. Rolfo, Godefridus J. Peters
1Department of Medical Oncology, VU University Medical Center, Amsterdam 1081 HV, the Netherlands.
2Department of Pathology, Antwerp University Hospital, Antwerp 2650, Belgium.
3Center for Oncological Research, University of Antwerp, Antwerp 2610, Belgium.
4Cancer Pharmacology Lab, AIRC Start-Up Unit, University of Pisa, Pisa 56124, Italy.
5Medical Oncology Unit, University Hospital of Parma, Parma 43126, Italy.
6University of Maryland Greenebaum Comprehensive Cancer Center, Baltimore, 21220 MD, USA.
Abstract Aberrant activation of the epidermal growth factor receptor (EGFR) is a driving force for cancer growth in a subgroup of non-small cell lung cancer patients. These patients can be identified by the presence of activating EGFR mutations.Currently three generations of EGFR-tyrosine kinase inhibitors (TKIs) have been approved by the Food and Drug Administration and European Medicine Agency. This paper reviews the structure of EGFR and the downstream signaling pathways of EGFR and describes the mechanisms of intrinsic and acquired resistance against EGFR-TKIs.These mechanisms include secondary or tertiary mutations in EGFR, the activation of bypassing signaling pathways or a histological transformation to small cell lung cancer. Moreover, drug efflux transporters will affect the cellular accumulation of EGFR-TKIs and penetration of the first generation of EGFR-TKI into the brain. Lysosomal sequestration of some EGFR-TKIs may also prevent the drugs to reach their target. In conclusion, resistance to EGFR-TKIs is multifactorial, including primary and acquired mutations in the EGFR gene, activation of bypassing pathways and limited uptake of drugs in the cells or target tissues. More pharmacological studies are needed in order to develop new specific compounds targeted to overcome new resistance mechanisms in order to enable a personalized treatment approach.
Keywords: Epidermal growth factor receptor, non-small cell lung cancer, resistance, EGFR-tyrosine kinase inhibitors,erlotinib, gefitinib, afatinib, osimertinib, rociletinib
INTRODUCTION
Molecular characterization of non-small cell lung cancer (NSCLC) and subsequent identification of targetable activated kinases led to a considerable shift in the treatment of this lethal disease in the past two decades. In patients with oncogene addicted NSCLC, particularly in adenocarcinoma, standard platinum-based chemotherapy has been replaced by molecularly driven strategies significantly improving the outcomes for these patients[1]. Among activated driver kinases suitable of pharmacological inhibition,epidermal growth factor receptor (EGFR) represents the most investigated and targeted one. Under physiological conditions, the EGFR signaling pathway controls cell growth, survival, proliferation, and differentiation by conveying the signal from the cell surface to downstream targets[2]. Oncogenic mutations in EGFR usually increase the kinase activity of EGFR, thus leading to hyperactivation of the pro-survival signaling pathway[3].
Approximately 50% of Asian patients and 11%-16% of patients from Western Countries with NSCLC harbor mutations in EGFR[4]. EGFR mutations are more often found in NSCLC from female never smokers,and tumor histology is mainly adenocarcinoma[5,6]. Three generations of specific EGFR-tyrosine kinase inhibitors (TKIs) have been developed and partially implemented in clinical practice for the treatment of EGFR-driven NSCLC up today: (1) gefitinib and erlotinib ( first-generation); (2) afatinib and dacomitinib(second-generation); and (3) osimertinib and rociletinib (third-generation)[1]. Despite initial response and tumor shrinkage, patients experience disease progression due to the onset of a resistance mechanism to targeted treatment. This review will provide a comprehensive overview of current knowledge about EGFR structure and EGFR mutations, focusing on resistance mechanisms to EGFR inhibition and additional strategies to overcome treatment resistance.
Structure and activation of wild-type EGFR
EGFR is a transmembrane receptor protein that belongs to the ErbB family of tyrosine kinases. EGFR is made up of an extracellular domain, also known as ligand binding region, a transmembrane region and an intracellular tyrosine kinase domain[7]. The EGFR kinase domain consists of two lobes: a C-terminal lobe(C-lobe) and an N-terminal lobe (N-lobe). The catalytic site is located in the cleft between these two lobes.As illustrated in Figure 1, the catalytic site contains the activation loop (A-loop) (contributed by the C-lobe), the regulatory C-helix and the nucleotide phosphate binding loop (P-loop) (both contributed by the N-lobe)[8].
For the activation of a tyrosine kinase receptor, two important conditions must be met. Firstly, the critical amino acids need to be positioned correctly to allow transfer of the phosphate group. Secondly, the peptide substrate binding site needs to be accessible[9]. In EGFR, the A-loop occludes the peptide substrate binding site. The C-loop contains a catalytic important glutamate that, together with Lys, is necessary for the coordination of the α- and β-phosphate groups on ATP[10]. The P-loop also contributes to the coordination of ATP through its GXGXXG motif[3].
In wild-type EGFR, the binding of EGF causes dimerization of the EGFR-receptor in a head-to-tail manner[11]. Hereby, the C-lobe of the one receptor pushes the N-lobe of the second receptor away, which leads to a reorientation of the A-loop. This leads to a translocation of the regulatory C-lobe to its active position[11], whereby Glu can interact with Lys. Furthermore, the peptide substrate binding site becomes accessible[10].
EGFR signaling
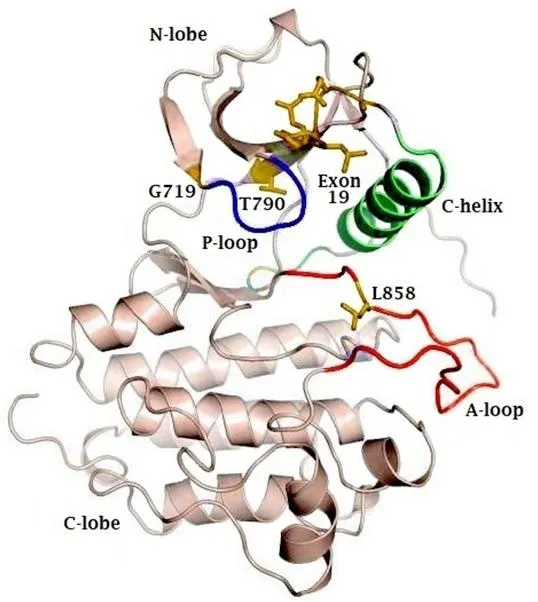
Figure 1. Representative figure of the crystal structure of the EGFR kinase domain, highlighting the N- and C-terminal lobes, as well as the phosphate coordinating P-loop (blue) and activation loop (A-loop). The active site lies in the cleft between the A-loop (red) and the C-lobe.The regulatory C-helix is colored in green. Sites of activating mutations are indicated in yellow. EGFR: epidermal growth factor receptor
After binding to its ligand, EGFR frequently heterodimerizes with its family members Her2, Her3 and Her4[12]. Eleven ligands are known within the family, of which EGF binds EGFR with the highest affinity[13].Signaling through EGFR can be divided in two main categories: kinase-dependent and kinase-independen signaling[14]. Kinase-dependent signaling results in the activation of several downstream pathways. A first activated pathway is Ras-Raf-MEK-ERK and JNK signaling[15,16], that promote cell survival and proliferation. A second signaling pathway is the activation of PI3K/Akt through the association of Her3[17].Thirdly, activation of EGFR also leads to STAT3 dimerization and translocation to the nucleus[18]. Fourthly,PLCγ signaling is activated through Src[19].
Besides kinase-dependent, EGFR also has kinase-independent functions. These include the stimulation of DNA synthesis[20], activation of mitogen-activated protein kinase (MAPK) signaling through heterodimerization[21], an anti-autophagic effect[22], modulation of protein subcellular trafficking (p53-upregulated modulator of apoptosis[23]) and activation of Akt through clustering in lipid rafts[24,25].
The influence of different mutations on EGFR activation
The vast majority of EGFR activating mutations (> 90%) are L858R point mutations and deletions within exon 19[26].
The residue L858 (exon 21) is part of a hydrophobic cluster on the A-loop. This cluster stabilizes the inactive conformation of EGFR through its hydrophobic interactions. The L858R mutation disrupts this hydrophobic interaction, which leads to destabilization of the inactive form and thus promotes the active conformation of EGFR[3,11]. The L861Q mutation is very close to the L858 residue, and most likely disturbs the inactive conformation in the same way[8].
Exon 19 deletion and exon 20 insertion take place in the C-helix. The exon 19 deletion in the loop of the C-helix “pulls” the regulatory helix in the active conformation. In contrast, the exon 20 insertion on the other end of the helix “pushes” the helix into its active conformation. Detailed crystal structures lack to confirm this hypothesis[3].
The G719 residue (exon 18) is a less frequent point mutation occurring in EGFR. This Glycine is located on the P-loop and is the first Gly in the GXGXXG motif. In the inactive form of EGFR, the P-loop contributes to the hydrophobic interactions that favor the inactive location of the C-helix, together with the cluster of L858. The G719 residue is necessary to cope with the torsion in the P-loop. Mutation to any other residue leads to a displacement of the P-loop, disturbs the hydrophobic interactions and thus destabilizes the inactive conformation of the receptor, resulting in receptor activation[3].
A study by Cho et al.[27]showed that dimerization of EGFR is not always necessary for downstream signaling. The wild-type EGFR receptor and the L858R mutant are dependent on dimerization for their activation, whereas in case of the exon 19 deletion, the exon 20 insertion and the L858R/T790M double mutant, dimerization is not necessary for EGFR-activation. This hypothesis is supported by studies with cetuximab, a monoclonal antibody that prevents EGFR dimerization. Cetuximab has been shown to inhibit EGFR-signaling most efficiently when the L858R mutation is present, whereas for the other mutations it only has a modest effect[27].
First- and second-generation EGFR-TKls
Several of the above mentioned mutations in EGFR render the tumor sensitive to EGFR-TKIs. Erlotinib and gefitinib are the first generation of EGFR-TKIs that were FDA and EMA-approved. Both erlotinib and gefitinib have anilinoquinazoline-based structures and serve as ATP-competitors[28]. The efficacy of both drugs is dependent on the type of sensitizing mutation[29]. Jiang et al.[29]observed a 100x increased sensitivity of the Ba/F3 variant with the L858R mutation to gefitinib, compared to a 6x increased sensitivity for the G719S mutant. This difference in sensitivity might be related to the increased affinity for ATP of both mutants. In comparison to wild-type EGFR, the L858R mutant has a 50x increased ATP-affinity,in contrast to a 10x increased affinity for the G719S mutant[29]. These differences in affinity and response might be related to the position of both mutants. As discussed above, the L858R mutation is located in the A-loop, whereas the G719S mutation is located in the P-loop of the receptor.
Although there is a difference in affinity of erlotinib and gefitinib for the mutant and wild-type EGFR receptors, the drugs will bind wild-type EGFR as well. This binding results in on-target side effects such as skin rash or diarrhea[30]. Regarding brain activity, the actual concentrations of erlotinib and gefitinib in the central nervous system are too low to be active due to activity of PGP and BCRP drug transporters[31,32].
Afatinib is a second-generation EGFR-TKI. The afatinib structure is also anilinequinazoline-based, and it inhibits both EGFR and ErbB family members Her2, Her3 and Her4. Afatinib is also able to penetrate the blood-brain-barrier (BBB)[33], but it showed moderate activity against brain metastasis. In contrast to the first generation EGFR-TKIs, afatinib binds the receptor irreversibly at the C797 residue[34]. This irreversible binding of the receptor often leads to more severe on-target side effects like skin rash and diarrhea. A meta-analysis on 3000 patients showed that afatinib has a limited effect on the L858R mutation, whereas it has higher efficacy against the exon 19 deletion[35,36]. The exon 20 insertion also leads to innate resistance against afatinib.
Moving to clinical implications of EGFR inhibition, two seminal studies firstly described responsiveness to gefitinib in EGFR-mutated NSCLC[37,38], opening a new landscape for the treatment of oncogeneaddicted lung cancer. Phase III trials have further explored the efficacy of first- and second-generation TKIs in this setting: a significant clinical benefit was observed for all the compounds in comparison with chemotherapy, in terms of progression-free survival (PFS), response rates and disease control rates. Indeed,the median PFS of first- and second-generation TKIs ranged from 8.0-13.1 months, which was consistent if compared to 4.6-6.9 months achieved by standard chemotherapy. Moreover, the benefit was independent of the line of treatment for which TKIs were administered, and no difference in efficacy was observed between first- and second-generation inhibitors in available comparative studies, except for the secondgeneration irreversible TKI dacomitinib[4]. These results allowed first- and second-generation EGFR-TKIs to become the new standard of care for frontline treatment of patients with EGFR-driven NSCLC.
Third-generation EGFR-TKls
Virtually all patients treated with first- and second-generation TKIs will acquire resistance to this targeted treatment within about one year. The onset of EGFR T790M secondary mutation is responsible for 50% of patients progression[39]. A more detailed description of all resistance mechanism is provided in the paragraphs below. Before the advent of third-generation EGFR-TKIs, patients who harbored T790M mutations had limited treatment options, which usually consisted of standard platinum-based chemotherapy. Even if afatinib demonstrated preclinical activity in T790M positive EGFR-mutated NSCLC[40], it showed no clinical benefit when given to patients who progressed after gefitinib or erlotinib[41].Development of compounds that can overcome T790M mutation was addressed by the introduction of third-generation EGFR-TKIs.
Osimertinib and rociletinib belong to the category of third-generation EGFR-TKIs. While osimertinib has been approved by the FDA and EMA, further development of rociletinib has been terminated[42].Osimertinib has a mono-anilino-pyrimidine core[43]and its binding is irreversible to the EGFR receptor via the C797 residue. It is active in the nanomolar range against both the sensitizing EGFR mutations and the T790M mutation. Like all the other inhibitors, it is not active against exon 20 insertions. Since both the insulin growth factor receptor 1 (IGF-1R) and the insulin receptor (IR) contain a gatekeeper, methionine,osimertinib is specifically designed not to inhibit both receptors, to prevent hyperglycemia in treated patients. This is in contrast to rociletinib, where the drug was intended to inhibit the IGF-1R and failed partially because of hyperglycemia[43]. Osimertinib is able to cross the BBB and shows activity against metastases in the central nervous system[44,45].
Since osimertinib was shown to have clinical activity in T790M mutated NSCLC after failure of previous TKIs in multiple studies[46-48], a study of T790M status is mandatory after progression to first- or secondgeneration inhibitors and it could consist of both blood and tumor tissue sampling analysis[49-51]. Indeed,after progression to gefitinib, erlotinib or afatinib, patients treated with osimertinib obtained a PFS of 10.4 months compared to 4.4 months achieved with the platinum plus pemetrexed doublet (HR 0.30, P < 0.001);the objective response rate (ORR) was 71% vs. 31% in the two arms, respectively[49]. Remarkably, osimertinib was superior to both erlotinib and gefitinib also when administered frontline: PFS was 18.9 months vs.10.2 months (HR 0.46; P < 0.001) for osimertinib and erlotinib/gefitinib, respectively, and the benefit was independent of T790M mutation[48]. Following these striking results, osimertinib has recently been approved by both the FDA and EMA for the first-line treatment of NSCLC with activating EGFR mutations,irrespective of T790M status[52].

Figure 2. Schematic overview of the signaling through EGFR and other receptor tyrosine kinases (e.g., cMET) underlying the connections within these pathways, and the main mechanisms of resistance (in red). EGFR: epidermal growth factor receptor; HGF: hepatocyte growth factor; EGF: epidermal growth factor; GAB1/GRB2: associated binding protein; GRB2: growth factor receptor bound protein 2; JAK: janus kinase; STAT: signal transducer and activator of transcription; PI3K: phospho-inositide3 kinase; Akt: -protein kinase B; mTOR: mammalian target of rapamycin; 4E-BP-1: eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1; S6K: ribosomal protein S6 kinase;PLCγ: phospholipase gamma; PIP2: phosphatidylinositol-4,5-biphosphate; IP3: inositol trisphosphate; Ras: Ras oncogene; Raf: Raf protooncogene; MEK: mitogen activated protein kinase; ERK: extracellular signal-regulated kinase
Resistance mechanisms to EGFR-TKls
Multiple mechanisms can cause primary and secondary resistance to EGFR-TKIs, as depicted in Figure 2.They could be schematically divided into five categories: (1) mutation within EGFR; (2) activation of bypassing pathways; (3) histological transformation; (4) aberrations in drug transporters; and (5) lysosomal sequestration.
Resistance mechanisms caused by EGFR mutations
Regarding primary resistance to EGFR-TKIs, the exon 20 insertion confers innate resistance to both reversible and irreversible inhibitors. The exon 20 insertion is a rare mutation in EGFR that occurs in approximately 4% of NSCLC, accounting for 5%-10% of all EGFR mutations in lung adenocarcinoma[53-55].The patients with exon 20 insertion included in clinical trials testing EGFR-TKI showed a very poor response in comparison with patients carrying an L858R mutation or an exon 19 deletion[54]. However,more recently poziotinib has been identified as an inhibitor of EGFR harboring the ex20 mutation. Due to its relatively small size and flexibility, poziotinib can circumvent steric changes induced by the ex20 insertion and is a potent inhibitor of EGFR ex20[56]. It is currently been tested in a phase II trial and initial data showed an ORR of 64% in 11 patients[55].
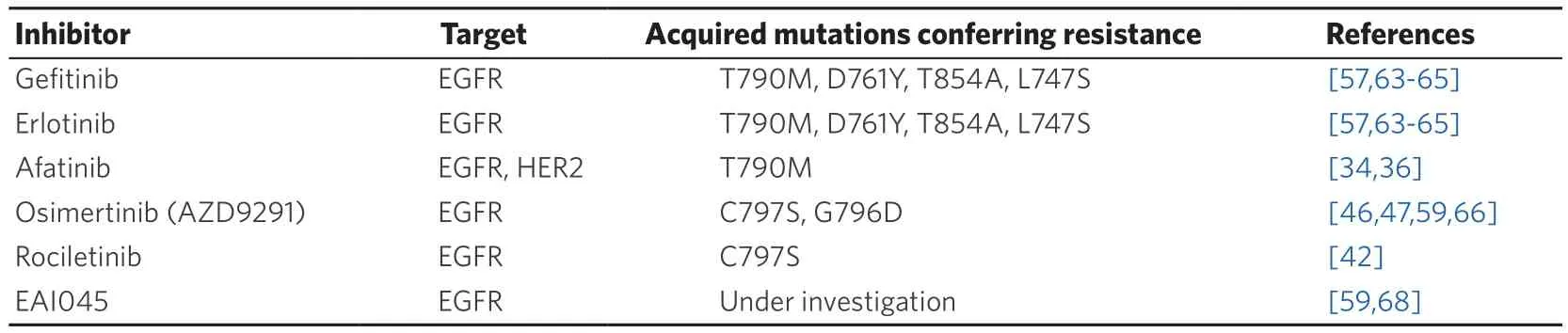
Table 1. EGFR-TKIs and EGFR mutations that confer drug resistance
Several mutations in EGFR are involved in secondary resistance to EGFR-TKIs, as summarized in Table 1.As stated above, 50%-60% of patients treated upfront with first- and second-generation EGFR-TKIs will experience a disease progression due to acquisition of a T790M mutation. The T790M mutation is located in the ATP binding cleft and is often referred to as “gatekeeper”-mutation[57]. However, crystal structures showed that the mutation does not sterically hinders the binding of the inhibitors. In contrast, it leads to a 4x-decreased affinity for gefitinib and an 18x increased affinity for ATP. This leads to an outcompeting of gefitinib by ATP. Moreover, the T790M mutation by itself stimulates the catalytic activity of the receptor,thus resulting in a survival advantage of these mutant cells[39].
The C797S mutation is a tertiary mutation that confers sensitivity to irreversible binding compounds like afatinib, osimertinib and rociletinib[58,59]. The C797S mutation targets the gatekeeper methionine residue that is used by the compounds to form a covalent bond with the receptor[60]. We need to discriminate between cells presenting with an L858R or exon 19 deletion in combination with the T790M and C797S mutation. Indeed, triple mutated cells are resistant to all generations of EGFR-TKIs; in this case, C797S mutation is associated to osimertinib resistance and occurs in 40% of patients treated with this thirdgeneration TKI[61]. On the other hand, cells containing an L858R or exon 19 deletion in combination with a C797S mutation alone, are still sensitive to the quinazoline-based inhibitors like erlotinib, gefitinib and afatinib[62]. Among rare EGFR mutations (< 1%), D761Y, T854A and L747S are responsible for acquired resistance to first-generation TKIs; however, the mechanism for how these mutations confer resistance is still unknown[63-66]. Of note, loss of T790M is another mechanism responsible for resistance to thirdgeneration osimertinib[67]. Fourth generation EGFR-TKIs able to overcome T790M and C797S are under development[68].
Resistance mechanisms mediated through bypassing pathways
Another mechanism for lung tumor cells to escape inhibition by EGFR-TKIs is the activation of parallel or downstream pathways. In this section, we will focus on best-known examples of pathways that can confer resistance to EGFR-TKIs. Activation of these pathways is not mutually exclusive with secondary or tertiary resistance mutations in EGFR[69]. Several of these bypassing pathways confer an intrinsic resistance to first-,second- and third-generation EGFR inhibitors, as well as to EGFR directed antibodies such as cetuximab.Some other pathways are activated during the course of treatment and can be considered as acquired resistance.
cMET
An important parallel pathway involved in resistance to EGFR inhibitors is the hepatocyte growth factor(HGF)-cMET pathway. The amplification of cMET plays a role in the activation of cMET-downstream signaling and thus circumvents EGFR-TKI inhibition. Amplification of cMET can be observed in approximately 3% of TKI-na?ve NSCLC cases, compared to 10%-20% of EGFR-TKI treated patients[69,70].The difference in percentage may represent acquired resistance. Secondly, when comparing downstream signaling of EGFR and cMET, these pathways overlap[71]. In vitro studies suggest a role in the reactivation of Her3/ERBB3-PI3K-Akt signaling[17]. This resistance mechanism is relevant for both the first- and thirdgeneration EGFR-TKIs[72-74].
KRAS
KRAS mutation represents another important intrinsic resistance mechanism to EGFR inhibition in NSCLC. KRAS mutations mainly occur in codon 12 (G12C/D/S/V) and codon 13 (G13C)[75]. KRAS is a downstream target of EGFR and normally becomes activated upon stimulation of EGFR. The mutations render KRAS constitutively active, thereby making KRAS independent of EGFR-stimulation. Therefore a patient harboring both sensitizing EGFR mutations concomitantly with KRAS-mutations is unlikely to respond to EGFR-TKIs[76], since EGFR-downstream signaling will continue its signaling due to active KRAS[77]. This has been reported for both the first and third generation of EGFR-TKIs[77,78].
Axl
Upregulation of the receptor tyrosine kinase Axl (anexelekto) is another intrinsic resistance mechanism against erlotinib and gefitinib. For the second- and third- generation EGFR-TKIs no reports on Axl as resistance mechanism are available. The resistance might be caused by either the upregulation of the Axl receptor, or the upregulation of its ligand GAS6[79]. Two effects can be observed that might explain the increased resistance against EGFR-TKIs. Firstly, Axl drives epithelial to mesenchymal transition(EMT). Hence, Axl downregulation leads to a decrease of the EMT markers N-cadherin and vimentin and the upregulation of E-cadherin[80]. Secondly, since PI3K-Akt and MAPK/ERK signaling cascades are downstream from both Axl and EGFR, increased activation of Axl can bypass EGFR-inhibition[81].
Her2
The Her2 receptor contains a kinase domain, but a ligand is still unknown. Her2 frequently dimerizes with EGFR and thus stimulates downstream signaling. Intrinsic aberrations in Her2 include overexpression,an insertion in exon 20 (G776YVMA) and amplification. It has been shown that, in case of Her2 overexpression[82]and Her2 amplification[83,84], EGFR-TKIs are still active in NSCLC patients. Despite of reports in cell line models and mouse models[85], EGFR-TKIs can be active against non-mutated Her2.Mutations in Her2 have been reported both in the kinase domain[86]and in the transmembrane domain[87],however, their concomitance with EGFR mutations is not clear thus far. All generations of EGFR-TKIs can inhibit non-mutant Her2[82,88,89]. Due to the heterodimerization of EGFR with Her2, stimulation of EGFR is often necessary to activate downstream signaling in case of non-mutant Her2. If EGFR is inhibited,downstream signaling is inhibited. In case of mutations in Her2, the receptor often gains in kinase activity and is no longer dependent on EGFR activation to initiate downstream signaling. However, these tumors show responses to Her2-inhibitors like, e.g., trastuzumab or lapatinib[86].
For colorectal cancer (CRC), the situation is different. In case of EGFR mutations in CRC the monoclonal anti-EGFR antibody cetuximab was the most effective treatment for patients. Since this antibody interferes with ligand binding to EGFR, the mechanism of action is different from the EGFR-TKIs administered in NSCLC, that block the kinase activity. Amplification of Her2 is a known resistance mechanism to cetuximab that can be overcome by combining cetuximab with Her2 inhibitors[90].
IGF-1R
The insulin growth factor receptor 1 (IGF-1R) has been shown to induce EMT in NSCLC cells carrying an EGFR exon 19 deletion[91]. In these cells, the activation of IGF-1R or stimulation with TGF-β resulted in a similar phenotype. Moreover, in reaction to EGFR-inhibition, it has been shown that increased EGFRIGF1R heterodimerization results in activation of mTOR, which in turn leads to de novo synthesis of EGFR and survivin[92,93]. Downstream signaling of IGF-1R results in increased resistance against EGFR-inhibitors.This mechanism of intrinsic resistance is observed with EGFR-TKI of the first[91-93], second[94]and third[95]generation. On the other hand, double inhibition of EGFR and IGF-1R at the same time can overcome this resistance[96,97].
FGFR
The fibroblast growth factor (FGF) family plays a role both in intrinsic and acquired resistance against EGFR-TKIs. In NSCLC, the ligands FGF2, FGF9 and the receptors FGFR1 and FGFR2 have been reported to be overexpressed[98,99]. Expression and activation of FGFR lead to a more mesenchymal phenotype and downstream signaling activates MEK-ERK and PI3K signaling[100]. Upon response to an EGFR inhibitor(erlotinib or gefitinib), FGF ligands and reporters become transcriptionally active[101]. Through the activation of an autocrine loop, this activation leads to EGFR-TKI resistance[102]. This resistance mechanism has also been reported for afatinib[103], but to date not for acquired resistance to osimertinib or rociletinib.
PIK3CA
The PIK3CA gene encodes the 110kDa catalytic subunit of PI3K[104], which is a downstream signaling pathway of EGFR. The PIK3CA pathway generally confers intrinsic resistance to first- and second-line EGFR inhibitors. In case of PIK3CA, both mutations and copy number gains are known aberrations.Mutations that increase the catalytic activity of PIK3CA occur in 1%-10% of EGFR-mutant NSCLC tumors[105,106]. Copy number gain (defined as > 2 copies per cell in over 40% of cells) is reported in approximately 40% of NSCLC patients[107]. Both mutations and copy number gain are correlated with a worse prognosis, overall survival and time to progression[105,107]. In case of EGFR-mutant tumors, 5/6 patients showed progressive disease when treated with EGFR-TKIs, 1 patient had treatment failure after only 4 months of treatment with EGFR-TKIs[105]. A meta-analysis summarized the effects of PIK3CA aberrations on treatment with diverse TKIs, amongst which are erlotinib, gefitinib and afatinib[108]. Reports on the effect of PIK3CA aberrations on osimertinib/rociletinib are not available yet.
BRAF
Overall, BRAF mutations can be considered as an intrinsic resistance mechanism but do not occur frequently in NSCLC. Indeed, BRAF mutations can be found in only 1.5%-3.5% of NSCLC patients and consists of a V600E mutation in about 50% of cases[109]. Concomitant mutations in EGFR and BRAF are even more rare, with reported percentages around or below 1% of EGFR mutant NSCLC patients,dependent on the study[110,111]. Crosstalk between BRAF and EGFR has been reported in a non-EGFR mutant context[112]. Hereby, the activation of BRAF results in the upregulation of EGFR ligands in a c-Jun dependent way. This results in the activation of EGFR-MAPK signaling[112]. It seems a logical step to assume that activation of BRAF can bypass EGFR inhibition. Indeed, case reports described patients harboring a BRAF V600E mutation during treatment with osimertinib[74,113]. In a peculiar case, a combination of osimertinib with the BRAF inhibitor encorafenib resulted in significant inhibition of cell growth.Unfortunately, the patient died before this combination treatment could be administered[113]. Nevertheless,this strategy can be extended to other cases of EGFR-TKI resistance through BRAF mutations.
PTEN
Phosphatase and tensin homologue (PTEN) is a tumor suppressor. Loss of PTEN is reported in about 5%of EGFR mutant NSCLC patients[106]and is linked to intrinsic resistance against EGFR-TKIs[114]. PTEN is a link between EGFR and Akt signaling. When PTEN is lost, EGFR signaling is uncoupled from Akt signaling. Inhibition of EGFR no longer results in inhibition of Akt signaling, resulting in resistance to EGFR-TKIs[115]. This has been demonstrated in cell lines, where resistance to erlotinib or gefitinib was correlated with PTEN levels. Overexpression of PTEN in cell lines with low/no PTEN levels resulted in increased sensitivity to erlotinib or gefitinib[114,115]. Loss of PTEN is both known as a primary mechanism of resistance (present in EGFR-TKI na?ve patients)[116]and as acquired mechanism of resistance[114]. For the latter, around 10% of patients without any other known resistance mechanisms are reported to have PTEN loss[114].
TP53
In NSCLC, TP53 mutations occur in approximately 50% of patients. This is also the case in EGFR-mutated NSCLC[117-119]. The in fluence of TP53 mutations on response and survival of TP53/EGFR double mutated patients is not clear from literature. One report showed a significantly lower progression free survival in case of TP53 exon 8 mutations and exon 19 deletions in EGFR[118]. However, two other studies reported no significant effect in TP53/EGFR concomitant mutated patients[117,119], so the role of TP53 mutations in EGFRTKI resistance is still unclear.
YAP
Yes-associated protein (YAP) is a part of the HIPPO-pathway, and seems to be associated with both intrinsic and acquired resistance to EGFR-TKI. Several links between EGFR-signaling and YAP have been reported. Firstly, activation of ERK1/2 is linked to activation with YAP. Depletion of ERK1/2 results in degradation of YAP, whereas ERK2 overexpression results in YAP rescue. The MEK1/2 inhibitor trametinib leads to decrease of YAP levels and decreased HIPPO-signaling[120]. Secondly, in cell lines with increased resistance against erlotinib, gefitinib or osimertinib, YAP is found in the nucleus (which is a sign of its activation)[121]. Overexpression of YAP leads to increased resistance to erlotinib[122]. YAP expression is also increased after EGFR-TKI treatment, both in cell line models and human NSCLC samples[123]. Moreover,activation of YAP leads to an increase in Axl expression and activation and a change to EMT-phenotype of the cells[123]. Knock-down of YAP results in resensitization of these cells to EGFR-TKIs. Inhibition of Axl activity also resensitizes to EGFR-TKIs[123].
NF-kB
The applause was so loud for the King of the jungle when he gave his call and swung on a curtain rope that no one seemed to notice me walk slowly to the center of the stage
Recently it was shown that acquired resistance to the novel third-generation EGFR-TKI rociletinib was mediated by activation of the NF-kB pathway[124]. This study focused on H1975 NSCLC cells, which harbor a T790M mutation, and are therefore resistant to gefitinib and erlotinib, but sensitive to rociletinib.Induction of resistance to rociletinib led to NF-kB activation replacing oncogenic EGFR signaling.Conversely, inhibition of this pathway with the proteasome inhibitor bortezomib sensitized the cells again to rociletinib. Since bortezomib showed some activity in drug combinations in NSCLC cells and patients[125-127], this approach seems promising as an alternative to sensitize patients progressive on treatment with third-generation EGFR-TKI.
In conclusion, there is a plethora of bypassing pathways to EGFR [Figure 2]. As our knowledge grows,more bypassing pathways will undoubtedly be discovered.
Resistance mechanisms through histological transformation
A peculiar resistance mechanism to EGFR-TKI is the histological transformation from NSCLC to small cell lung cancer (SCLC), which occurs in approximately 14% of cases[128]. This has been reported for the first time in 2006 by Zakowski et al.[129]. The “transformed” tumor retains the EGFR mutation, but is no longer responsive to EGFR-TKIs[130]. Although several cases of SCLC have been reported, a lot of questions remain on the mechanism and origin of this transformation. However, recent genetic studies[131,132]revealed that all transformed SCLC have an inactivation of RB1 and TP53 in common. Since the treatment regimens for NSCLC and SCLC are very different, this raises some questions about the optimal therapy in the occurrence of a histological transformation. Four patients with a SCLC transformation were switched to a SCLC-oriented platinum-etoposide chemotherapy, which produced marked responses in three patients[128]. Moreover, case reports show good results from “drug-holidays”. For example, a patient originally presenting with an L858R positive adenocarcinoma showed a good initial response to erlotinib.When resistance occurred, a rebiopsy showed a SCLC subtype containing both an L858R and a PIK3CA mutation. This SCLC was resistant to erlotinib. After a couple of months without any EGFR-TKI the tumor had switched back to the adenocarcinoma subtype, retained the L858R mutation but the PIK3CA mutation could not be detected anymore. After a couple of months of response to erlotinib, the patient became resistant again, and rebiopsy showed again the SCLC subtype with both an L858R and PIK3CA mutation[128].
Resistance through aberrations in drug transporters
Several families of drug transporters are currently known which may play a role in resistance to EGFR inhibitors in NSCLC[133,134]. Examples are the organic anion transporters, organic cation transporters (OCT),organic anion transporting polypeptides, equilibrative nucleoside transporters and the ABC-transporter superfamily which contains the Breast Cancer Resistance Protein (BCRP), P-glycoprotein (PGP) and multidrug resistance protein (MRP) family[134]. Gefitinib uptake seems mediated by an OCT and the uptake of erlotinib seems to have a mixed pattern, active and passive. However, literature is not consistent because of the great diversity of drug transporters and their functions on TKIs.
Erlotinib and gefitinib both have been shown to interact with ABCB1/PGP and ABCG2/BCRP; one interesting aspect is their role on trafficking of the transporters, affecting their function[135]. The in fluence of these efflux pumps has been studied most extensively in relation to gefitinib. Gefitinib acts both as a substrate and as an inhibitor of these pumps. Hereby, the concentration of gefitinib is important. At low concentrations (IC50values in nmol/L range) gefitinib activates BCRP-ATPase activity in cell lines,whereas at concentrations > 1 μmol/L this stimulatory effect is lower[136,137]. Transduction of BCRP in gefitinib sensitive cell lines in the nanomolar range lead to gefitinib resistance. The BCRP-inhibitor Ko143 could reverse this resistance. However, this effect was much less in cell lines that are relatively insensitive to gefitinib (IC50values in μmol/L range)[138]. Due to its role as a BCRP-inhibitor, gefitinib is also able to overcome BCRP-mediated resistance in vitro[138,139].
Erlotinib and efflux pumps are studied less well. Erlotinib is a target of PGP and BCRP, but not of MRP2[140]. In the triple knock-out mouse (BCRP1/MDR1a/MDR1b-/-), the oral bioavailability of erlotinib was increased with 66%, which led to increased systemic exposure to erlotinib[140], but in cell lines overexpressing BCRP the sensitivity to erlotinib could not be increased by specific inhibitors of BCRP such as Ko143, questioning the role of BCRP[141].
Afatinib is both a substrate and inhibitor for the efflux pumps PGP and BCRP, however, the therapeutic doses of afatinib are too low to affect the function of these pumps in vivo[142].
Lysosomal sequestration
Lipophilic or amphiphilic compounds can undergo a process called lysosomal sequestration,lysosomotropism or acid trapping[143]. These compounds are characterized by a pKa between 6.5 and 11[144].Due to this pKa the compounds are neutral in the cytosol, which has a pH of around 7.2-7.4, and can diffuse passively through the membranes. When crossing the membrane of the lysosomes, the pH lowers to around 4.5-5. Hereby the compounds become protonated in the lysosomes and can no longer diffuse through the membranes and become “trapped” in the lysosomes[145].
Lysosomal trapping has been identified as a resistance mechanism against anticancer drugs by shielding the respective targets from the drugs[148,149]. Furthermore, this drug accumulation causes lysosomal stress, resulting in the activation of the coordinated lysosomal expression and regulation-pathway by transcription factor EB[145]and in lysosomal exocytosis[150].
Focusing on the EGFR-TKIs: erlotinib does not accumulate in the lysosomes by lysosomal sequestration[134],gefitinib on the other hand is a lysosomotropic compound[143]. As mentioned above gefitinib's uptake is mediated by an active process and subsequently the drug seems to be trapped in the cells[151]. In contrast,erlotinib is likely to be transported into the membrane, binds to the target and is effluxed before entering the cell[151]. For the second- and third-generation EGFR-TKIs, this needs to be assessed.
Future directions
The ultimate goal of every anticancer treatment is to cure cancer, by delaying disease progression and providing a longer overall survival. Unfortunately, in the case of advanced NSCLC, despite the giant steps that have been made in the last decade, a definitive cure is still far from being discovered. The majority of the NSCLC patients lack a targetable driving mutation, and in this case they could be treated with immune checkpoint inhibitors or combinations of cytotoxic drugs, such as gemcitabine-cisplatin (or carboplatin),gemcitabine and a taxane, a taxane and cisplatin (or carboplatin), pemetrexed and cisplatin (for nonsquamous NSCLC)[1,152]. Although these combinations have been extended with TKIs, the approach was usually not successful. Therefore, concerning EGFR-mutated NSCLC, a current challenge is to stay ahead of resistance against EGFR-TKIs.
A first strategy is to simultaneously inhibit multiple signaling pathways with combination therapy, such as a dual inhibition of EGFR and cMET signaling. This can prevent the activation of the bypassing cMET pathway upon EGFR inhibition and thus block a possible way-out for the cancer cells. However, given the multiple possible escape strategies, it remains a big challenge to predict the future mechanism of resistance of a specific tumor and target it from the beginning. Moreover, in order to be able to determine the acquired mechanism of resistance, continuous research is needed. Once a novel mechanism of resistance is discovered it is necessary to expand the screening strategies in clinical routine to bring it to the patients.However, sometimes the discovered mechanism of resistance is not fully targetable. A good example is the occurrence of mutations in KRAS: although extensive research has been conducted into Ras-inhibitors, no suitable drug is available to date.
A second strategy is a combination with immunotherapy, although an extensive discussion of these combinations is beyond the scope of this paper. In NSCLC, immunotherapy seems most effective in patients with a high tumor load, but all predictive parameters are still under investigation and need to be validated. A remaining question will be whether immunotherapy should be given before treatment with a TKI or whether the TKI should be given until resistance and then be followed with immunotherapy[153].Alternatively, TKIs might be combined with immunotherapy, but this approach is in its infancy.However, certain types of therapy, including standard chemotherapy and some TKIs can induce synthesis and excretion of several cytokines and chemokines, which play an important role in the efficacy of immunother
A third strategy is to reconsider combinations of anti-signaling drugs (including anti-angiogenesis drugs) with standard therapies such as gemcitabine, pemetrexed, cisplatin, taxanes and radiation. These combinations should be designed taking into account the effect of these standard therapies on signaling pathways and the tumor surroundings. These drugs usually activate the AKT pathway as a survival mechanism[154]. Both in experimental systems and in patients it has been demonstrated that inhibition of AKT or EGFR can neutralize the AKT activation and enhance efficacy of standard drugs. For instance in several NSCLC models erlotinib increased the efficacy of pemetrexed[155], while inhibition of AKT with perifosine enhanced the efficacy of the platinum analog satraplatin[156]. MK2206 enhanced the effect of radiation alone[157]or in combination with gemcitabine and radiation (El-Naggar, unpublished results).Unfortunately, the current AKT inhibitors (e.g., MK2206, perifosine) were not sufficiently effective in patients, but several novel inhibitors are under development.
Finally, the implementation of liquid biopsies can be helpful, both for monitoring, e.g., the percentage of mutated cells in a tumor over time and for screening the occurrence of mechanisms of resistance during the treatment (as already approved for T790M detection). This approach can provide the treating physicians with the necessary information to decide to either continue treatment or to switch to another strategy in time (at the earliest sign of resistance).
The in fluence of drug efflux transporters and lysosomal sequestration on the intracellular concentration and location of small molecule inhibitors should not be overlooked. This is illustrated with erlotinib and gefitinib, which do not reach therapeutic concentrations in the brain, because of rapid efflux. Since osimertinib readily passes the blood-brain barrier and is not effluxed, patients with brain metastases should receive first-line treatment with osimertinib.
CONCLUSION
Identification of EGFR mutations in NSCLC and development of targeted treatment led to a revolution in the treatment of this peculiar oncogene-driven disease. The TKIs erlotinib, gefitinib, afatinib and osimertinib have been implemented in current clinical practice. However, the successful inhibition of EGFR signaling pathway can be limited by different mechanisms of resistance. A very important role is played by the T790M and C797S mutations that arise in most patients after treatment with EGFR-TKIs.Moreover, the identification of additional resistance mechanisms by activating bypassing pathways has taught us a lot on the intertwining of intracellular signaling pathways and gives us more possibilities to overcome this resistance by inhibiting multiple pathways simultaneously.
The activity of TKIs can also be influenced by other resistance mechanisms such as histological transformation, drug efflux transporters and lysosomal sequestration. The latter mechanisms need to be taken into account for the development and testing of new TKIs, in order to prevent lower than expected activity of these drugs in the clinical practice.
DECLARATIONS
Authors' contributions
Original concept and writing: Van Der Steen N
Supervision and revision: Giovannetti E, Peters GJ
Writing and revision: Carbone D, Leonetti A
Supervision: Rolfo CD
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
? The Author(s) 2018.
- Cancer Drug Resistance的其它文章
- Meeting Abstracts of the BACR conference:response and resistance in cancer therapy
- What really matters - response and resistance in cancer therapy
- Wilms' tumor gene (WT1) is strongly expressed in high-risk subsets of pediatric acute lymphoblastic leukemia
- Epigenetics, a key player of immunotherapy resistance
- Antibody drug conjugate development in gastrointestinal cancers: hopes and hurdles from clinical trials
- Acknowledgement to reviewers of Cancer Drug Resistance in 2018