
Rapid and sensitive analysis of melatonin by LC-MS/MS and its application to pharmacokinetic study in dogs
2017-01-19 06:26:04
School of Pharmacy,Shenyang Pharmaceutical University,Shenyang,China
Rapid and sensitive analysis of melatonin by LC-MS/MS and its application to pharmacokinetic study in dogs
Huimin Zhao,Yifei Wang,Yi Jin,Shu Liu,Haiyan Xu,Xiumei Lu*
School of Pharmacy,Shenyang Pharmaceutical University,Shenyang,China
A R T I C L EI N F O
Article history:
Received 16 June 2015
Received in revised form 12 August 2015
Accepted 16 August 2015
Available online 24 August 2015
Melatonin
A rapid and sensitive liquid chromatography tandem mass spectrometry method was established and validated for determination of melatonin in dog plasma using desvenlafaxine as an internal standard(IS).Plasma samples were pretreated by liquid–liquid extraction with ethyl acetate.Chromatographic separation was carried out on a C18 column at a fow rate of 0.2 ml/min by an isocratic mobile phase of methanol:5 mM ammonium acetate:formic acid(40:60:0.1,v/v/v).Positive ion mode detection was performed using multiple reaction monitoring(MRM)at m/z 233.2→174.2 for melatonin and m/z 264.2→58.2 for desvenlafaxine. The method was linear in the concentration range of 0.020–10 ng/ml with a correlation coeffcient≥0.996.The intra-and inter-assay precision(%RSD)values were within 12.6%(LLOQ 15.2%),and accuracy(%RE)ranged from?1.8%to 5.0%(LLOQ±16.5%).The total analysis time was 3.0 min.The method was fully validated and successfully applied to a pharmacokinetic study of melatonin prolonged-release tablet in Beagle dogs.The values of half-life and
Tmaxwere similar to the corresponding data reported before.
?2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license
(http://creativecommons.org/licenses/by-nc-nd/4.0/).
1.Introduction
Melatonin(N-acetyl-3-(2-aminoethyl)-5-methoxyindole),an endogenous hormone,is a predominant product of the pineal gland.It is secreted in a rhythm that is strictly dependent on the light–dark cycle.The melatonin plasma concentration rises in the early night,peaks at about midnight and then declines during the daytime[1].Melatonin also has an age-related rhythm.The reduction of melatonin contributes to the aging process and may shorten the life span[2].Studies have shown that exogenous melatonin administration is useful for treating circadian disruptions,e.g.,insomnia[3],jet lag[4].Moreover, melatonin has pharmacological effects on the treatment of Alzheimer’s disease[5],Parkinson disease[6],glaucoma[7], depressive disorder[8],breast[9]and prostate cancer[10],hepatoma[11]and melanoma[12].Unfortunately,a substitution therapy is not easily achieved with melatonin because of its relatively poor bioavailability[13]and rapid elimination[14]. In 2007,a prolonged-release formulation of melatonin(Circadin?,Neurim Pharmaceuticals,Tel-Aviv,Israel)was approved in Europe by the European Medicines Agency(EMEA) for the treatment of insomnia in patients aged>55 years[15]. Prolonged-release formulation is often used in clinical treatment to maintain longer effective concentrations and lower incidences of side reactions.
Several methods for the determination of melatonin in biological fuids have been reported,such as enzyme immunoassay (EIA)[16],radioimmunoassay(RIA)[17],high-performance liquid chromatography with fuorescence[18–20]or electrochemical detection[21–23],capillary electrophoresis-ultraviolet detection(CE-UV)[24],gas chromatography–mass spectrometry(GC-MS/MS)[25–29]and liquid chromatography coupled with tandem mass spectrometry(LC-MS/MS)[30–41].Perhaps immunoassay is widely used in the determination of melatonin,but even so,it has the disadvantage of cross reactivity affected by structurally similar compounds.HPLC coupled with fuorescence or electrochemical detection,CE-UV or GC-MS was not adequate to perform a pharmacokinetic study of melatonin because of its complex sample preparation procedure while LC-MS/MS demonstrates great selectivity and relatively good sensitivity.Wang et al.established an LC-MS/MS method to determine melatonin in dog plasma with an LLOQ of 0.001 ng/ml, whereas it suffered from tedious sample extraction,extravagant plasma of 0.55 ml and long analytical time(11 min)[35]. Yang et al.developed an LC-MS/MS method using liquid–liquid extraction with an LLOQ of 0.01 ng/ml[37].However,a bulk plasma volume of 0.5 ml and a large injection volume of 20 μl were used in this method and the analytical time was about 6 min.
In this paper,we developed a rapid and sensitive LC-MS/ MS method to determine melatonin in dog plasma.The method was greatly suitable for high throughput analysis and was successfully applied to a pharmacokinetic study of melatonin in Beagle dogs after an oral dose of 2.0 mg prolonged-release melatonin tablet.
2.Materials and methods
2.1.Chemicals and reagents
Melatonin(99.5%purity)and desvenlafaxine succinate(used as IS,100.8%purity)were obtained fromWuhan Xingjialing Biotech Co.Ltd(Wuhan,China)and Shenzhen Jianzhu Bio-tech Co.Ltd(Jinan,China),respectively.Melatonin prolongedrelease tablets(2 mg/tablet,Lot:457026302,RAD Neurim Pharmaceuticals EEC Ltd.,Israel)were a gift from Beijing Boaiwangkang Bio-tech Co.Ltd(Beijing,China).Methanol(HPLC grade)was purchased from DikmaTechnology(Lake Forest,CA, USA).Formic acid(HPLC grade)was purchased from ANPEL Scientifc Instrument Co.Ltd(Shanghai,China).Ethyl acetate, ammonium acetate and anhydrous sodium carbonate(analytical grade)were purchased from Tianjin Damao Chemical Technology Co.Ltd(Tianjin,China).Purifed water was purchased from Wahaha Co.Ltd(Hangzhou,China)and fltered through a 0.22 μm membrane flter before use.
2.1.1.Chromatographic and mass spectrometric conditions
Chromatographic separation was accomplished on a Luna C18 column(2.0×50 mm,3 μm,Phenomenex Technology,USA). HPLC analysis was performed on an Agilent 1290 LC system (Agilent,Santa Clara,CA,USA)equipped with a bin pump (G4220A),an auto-sampler(G4226A)and a thermostated column compartment(G1316C).An isocratic mobile phase of methanol:5 mM ammonium acetate:formic acid(40:60:0.1,v/v/v)was applied at a fow rate of 0.20 ml/min.The injection volume was 10 μl.The temperatures of the column and auto-sampler were kept at 35°C and 4°C,respectively.
Mass spectrometry detection was carried out on an AB Sciex (Applied Biosystem/MDS SCIEX,Foster City,CA,USA)API4000 mass spectrometer,using TurboionsprayTMsource in positive ion mode.Quantifcation was performed using multiple reaction monitoring(MRM)mode with the transitions of m/z 233.2→174.2 for melatonin and m/z 264.2→58.2 for desvenlafaxine.The optimal source parameters were as follows: collision-activated dissociation gas(CAD)6 psi,curtain gas(CUR) 30 psi,nebulizer gas(gas 1)50 psi,heater gas(gas 2)50 psi,ion spray voltage 5500 V and source temperature 500°C.The compound dependent parameters collision energy(CE)was set at 19 V for melatonin and 39 V for IS.Declustering potentials(DP) for melatonin and IS were 60 and 52 V,respectively.Entrance potential(EP)and cell exit potential(CXP)for both melatonin and IS were maintained at 10 and 15 V,respectively.Quadrupole 1 and quadrupole 3 were kept at unit resolution and dwell time was set at 100 ms.System control and data analysis was performed by AB Sciex Analyst software(version 1.5.2).
2.2.Preparation of standard solution,calibration curve and quality control samples
Stock standard solutions of melatonin and desvenlafaxine(IS) were prepared in methanol at a concentration of 1.0 mg/ml. A series of melatonin working standard solutions with concentrations in the range of 0.20–100 ng/ml were obtained by further dilution of the stock solution with methanol.The IS working solution was diluted with methanol to a fnal concentration of 12.5 ng/ml.All solutions were stored at?20°C and brought to room temperature before use.
The plasma calibration standards of melatonin with fnal concentrations of 0.020,0.050,0.20,1.0,5.0,10 ng/ml were prepared as follows:a 20 μl aliquot of melatonin working standard solutions was spiked into a 10 ml glass tube and the solvent was evaporated to dryness under air stream.A 200 μl aliquot of blank plasma was then added to each tube.Quality control (QC)samples of 0.040,0.40 and 8.0 ng/ml were prepared in the same way as calibration standards.Additional validation QCs were prepared at 0.020 ng/ml to test LLOQ.The spiked plasma (calibrators and QCs)were treated according to the sample preparation described below.
2.3.Sample preparation
To a 200 μl aliquot of each plasma sample,20 μl of IS solution (12.5 ng/ml desvenlafaxine in methanol)and 50 μl of 1 M sodium carbonate were added and vortex-mixed for 10 s.After the addition of 1 ml of ethyl acetate and 100 μl of water,the sample was shaken on a reciprocating shaker at 240 circles per minute for 10 min,followed by centrifugation at 4250×g for 5 min.The organic layer was separated and evaporated to dryness at 40°C under an air stream.The residue was reconstituted in 100 μlof the solution of methanol:5 mM ammonium acetate(40:60, v/v)and 10 μl was injected for LC-MS/MS analysis.
2.4.Method validationA full validation process was conducted according to the Guidance for Industry Bioanalytical Method Validation,recommended by FDA[42].
Selectivity of the method was evaluated by analyzing six blank plasma samples to investigate the potential interferences at the retention times for the analyte and IS.Carry over was carried out by injecting blank plasma samples after the injection of the upper limit of quantifcation(ULOQ)sample (10 ng/ml).Cross-talk of multiple reactions monitoring(MRM) for the analyte and IS was checked using the highest standard on calibration curve and working solution of IS.
Linearity of the method was assessed by processing(in duplicate)a six-point calibration curve over the concentration range of 0.020–10 ng/ml.Calibration curves were built by plotting the peak area ratios of the analyte to IS versus the nominal concentrations of the analyte with weighted(1/x2)leastsquares linear regression.The correlation coeffcient(r)of a calibration curve should be more than 0.99.The lowest limit of quantifcation(LLOQ)was defned as the lowest concentration on the calibration curve with a signal-to-noise ratio of at least 10.The back-calculated concentrations at each point have to be within±15%deviation from the nominal value except for LLOQ where deviation should be within±20%.
Intra-day and inter-day precision and accuracy were determined by analyzing six replicate QCs at plasma concentrations of 0.020(LLOQ),0.040,0.40 and 8.0 ng/ml on the same day and on three consecutive days,respectively.The criteria for the data included accuracy(relative error,RE)within±15% (±20%for LLOQ)and a precision(relative standard deviation, RSD)not exceeding 15%(20%for LLOQ).
The extraction recovery and matrix effect of melatonin and IS were determined by analyzing six replicates of plasma samples at three QC levels of 0.040,0.40 and 8.0 ng/ml of melatonin and IS concentration of 1.25 ng/ml.The recovery was calculated by comparing the peak areas obtained from extracted spiked samples with the mean peak area of samples spiked post-extraction at corresponding concentrations.The matrix effect was measured by comparing the peak areas of analyte added into post-extracted blank plasma samples with the mean peak area obtained from corresponding standard solution of the analyte prepared in the reconstitution solution. The variability(RSD)of the matrix effect at each concentration should be less than 15%[43].
The stability of melatonin in dog plasma was assessed by analyzing triplicates of QCs at 0.040 and 8.0 ng/ml under different storage conditions.These QCs were analyzed after storage at room temperature for 4.0 h(bench-top stability),at?70°C for 25 d(long-term stability),after three freeze–thaw cycles at?70°C(freeze–thaw stability),after storage with dry ice for 2.0 h (transportation stability),and in processed samples at 4°C for 24 h(auto-sampler stability).
The concept of incurred sample reanalysis(ISR)was frst introduced in the 2007 White Paper[44].And then,running an ISR was recommended in the 2011 European Medicines Agency Guidance and the 2013 FDA Guideline Draft.Based on these guidelines,15%of the samples from six different dogs atCmax, the time of absorption and elimination phase in pharmacokinetic profle were prepared.Two-thirds(67%)of the repeated sample results should be within 20%,which is determined with the following equation:RE%=(Repeat?Original)/Mean*100%.
2.5.Pharmacokinetic study
Six Beagle dogs(10.10±1.10 kg,three male and three female) were supplied by Shenyang Kangping Laboratory Animal Institute(Shenyang,China).They were housed under standard conditions and hadad libitumaccess to water.All experimental procedures were performed in accordance with the guidelines of the Experimental Animal Care and Use Committee of Shenyang Pharmaceutical University(Shenyang,China). Dogs were given an oral dose of 2.0 mg melatonin prolongedrelease tablets 2 h after dinner.Blood samples(1.0 ml)were collected into heparinized tubes before(0 h)and at 0.17,0.33, 0.5,1.0,1.5,2.0,2.5,3.0,4.0,5.0,6.0,8.0,10 and 12 h after administration.To investigate whether the endogenous melatonin interfered with the determination or not,1.0 ml of“blank”blood sample was collected in accordance with the blood collection point as administration on the previous day.Plasma was immediately separated by centrifugation and stored at?70°C until analysis.
Pharmacokinetic parameters of melatonin were calculated by non-compartmental method using DAS 3.2 pharmacokinetic program(Chinese Pharmacology Society).The maximum plasma concentrations(Cmax)and the corresponding peak time (Tmax)were observed directly from the individual drug plasma concentration–time profle.The terminal elimination rate constant(ke)was estimated by log-linear regression of concentrations observed during the terminal phase of elimination. The elimination half-life(t1/2)was calculated as 0.693/ke.The area under the plasma concentration–time curve was calculated by the linear trapezoidal rule.
3.Results and discussion
3.1.Method development
3.1.1.Liquid chromatography
Various mobile phase conditions were evaluated in the experiment to obtain optimized responses,suitable retention times,and good peak shapes.Compared with water,5 mM ammonium acetate signifcantly made the shape of melatonin more symmetric.Formic acid(0.1%)increased the signal response of melatonin.Methanol was found to produce better elution ability than acetonitrile.Finally,a mobile phase consisting of methanol:5 mM ammonium acetate:formic acid (40:60:0.1,v/v/v)was chosen to obtain satisfactory sensitivity and good retention of melatonin.Under the optimized LC conditions,the retention times of melatonin and the IS were 2.6 and 1.3 min,respectively.The total analytical time was 3.0 min per sample.It was the shortest analysis time for analyzing melatonin in dog plasma compared to the research published before which was greatly suitable for high throughput analysis in pharmacokinetic studies.We also reduce the amount of methanolin mobile phase by applying a small particle size C18 column (2.0×50 mm,3 μm)on chromatographic separation at a fow rate of 0.2 ml/min.
3.1.2.Mass spectrometry
In our study,the LC-MS/MS analysis was performed in both positive and negative ion ESI modes.The positive ion mode was more suitable for quantitative determination because of its better signal-to-noise ratios.Melatonin was easily ionized in Turboionspray(TIS)interface to form protonated ion of[M+H]+at m/z 233.2.The product ion scan spectra of[M+H]+showed several major fragment ions at m/z 174.2 and 216.2.Following detailed optimization of MS conditions,fragment ion of melatonin at m/z 174.2 gave higher signal response and lower noise level than fragment ion at m/z 216.2.Hence ion transition of m/z 233.2→174.2 was fnally chosen for quantifcation of melatonin.Similarly,mass transition of m/z 264.2→58.2 was used for detection of desvenlafaxine(IS),which showed similar extraction behavior and signal drift trend with melatonin. Desvenlafaxine is a kind of selective serotonin reuptake inhibitor(SSRI)class antidepressant.Uz et al.showed that the SSRI had a positive effect on arylalkylamine-N-acetyltransferase gene expression and might modulate melatonin synthesis,but it only worked in the hippocampus and striatum[45].In this experiment,desvenlafaxine was simply added into the plasma in the process of sample preparation which would not have interaction with melatonin.So in this study,desvenlafaxine was chosen as the IS for melatonin.The product ion mass spectra of melatonin and IS are shown in Fig.1.
3.2.Sample preparation
Protein precipitation and liquid–liquid extraction(LLE)were applied and compared for the sample preparation.Marked suppression matrix effect was obtained after protein precipitation. Liquid–liquid extraction is a more effective method to avoid matrix effect,thus it was adopted in the present method for the preparation of samples.Diethyl ether and ethyl acetate were investigated to identify the more effcient extraction agent for the sample.To further improve the recovery of the analyte, sodium carbonate and sodium hydroxide as the alkaline reagents at different concentrations were considered.The resultshowed that ethyl acetate and 1 M sodium carbonate offered better extraction recovery.In this method,only 200 μl of dog plasma was used in each sample which was enough for the pharmacokinetic study of melatonin.
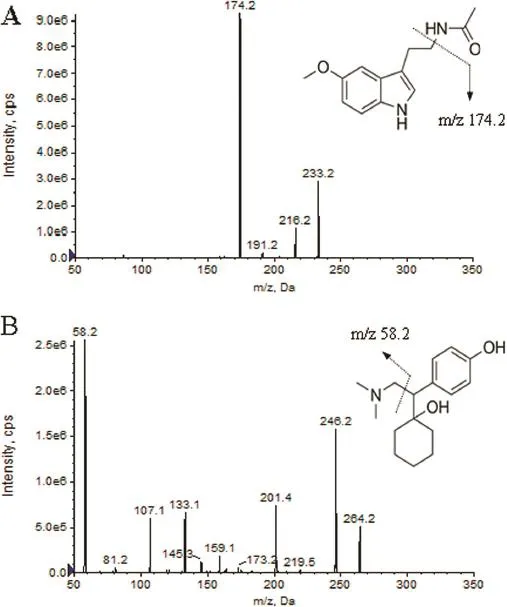
Fig.1–Production scan spectra of melatonin(A)and desvenlafaxine(IS,B).
3.3.Method validation
3.3.1.Selectivity,carry over and cross-talk
There was no evidence of interfering peaks from endogenous compounds at the retention times of melatonin and IS in all the tested six lots of blank matrix samples.The peak detected in the plasma collected on the previous day before administration was far less than 20%of the LLOQ.No carry over peaks were shown after the injection of ULOQ.No cross-talk peaks were observed between melatonin and IS after the injection of the highest standard on calibration curve and working solution of IS.Representative chromatograms obtained from the blank plasma collected at 01:00 AM on the previous day before administration,blank plasma spiked with melatonin at the LLOQ and a dog plasma sample taken at 2 h post-dose are shown in Fig.2.
3.3.2.Linearity and LLOQ
The linearity was evaluated on three separate days with two sets of calibration curves per day.The standard curve was constructedtospanananalyticalmeasuringrangeof0.020–10 ng/ml with good reproducibility and linearity.A typical equation of the calibration curve on a validation batch was as follows:y=0.231x?0.000206,whereyrepresents the peak area ratio of melatonin to IS andxrepresents the plasma concentration of melatonin.The correlation coeffcients of the generated calibrationcurvesweregreaterthan0.996.TheLLOQwasconfrmed to be 0.020 ng/ml,at which the accuracy was within±16.5%and the precision was within 15.2%(Table 1).
Yang et al.developed an LC-MS/MS method with an LLOQ of 0.01 ng/ml[37].The sensitivity of our method seemed not as good as theirs.However,our method was in fact more sensitive than the reported method.In the published method,Yang et al.used a larger plasma volume of 0.5 ml and an injection volume of 20 μl[37].In the present study,a 0.2 ml aliquot of plasma and an injection volume of 10 μl were used,which resulted in absolute sensitivity that was 2.5 times higher than the reported method.The sensitivity of this method could be easily improved by increasing the plasma volume and injection volume.However,this strategy was plasma wasted and might decrease dogs’tolerance.In addition,single dose administration of 2.0 mg melatonin resulted in aCmaxapproximately at 0.40 ng/ml[35].Therefore,the method with an LLOQ of 0.020 ng/ml was sensitive enough for pharmacokinetic study of melatonin.
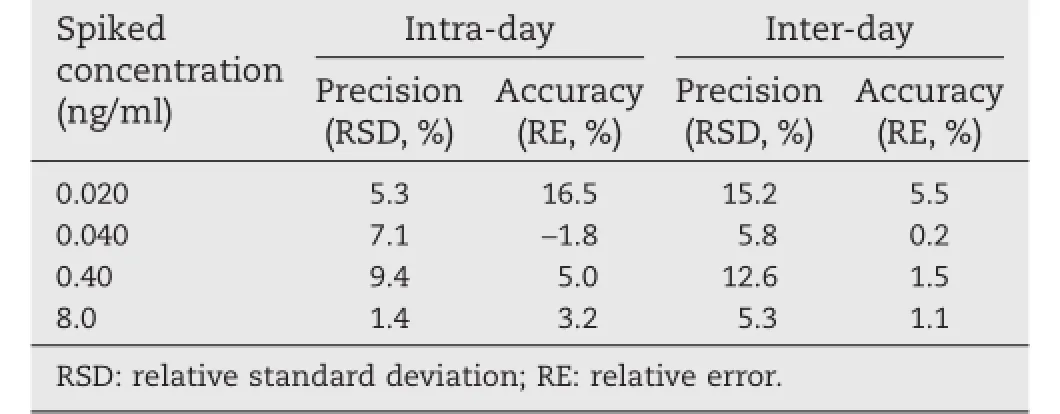
Table 1–Intra-and inter-day precision and accuracy of the LC-MS/MS method to determine melatonin in dog plasma(n=3 d,6 replicates per day).
3.3.3.Precision and accuracy
The results of intra-day and inter-day precision and accuracy of the assays are shown in Table 1.The intra-and inter-day precisions were all below 15%with a maximum RSD of 12.6%, and accuracy was calculated within 98.2%–105.0%.
3.3.4.Extraction recovery and matrix effect
The recoveries of melatonin extracted from dog plasma were 79.0±6.4,83.9±4.5 and 79.1±1.2%at the concentrations of 0.04, 0.40 and 8 ng/ml,respectively.The recovery of IS was 90.7±8.1%. The extraction recoveries of melatonin and IS were consistent and reproducible.The matrix effects of melatonin from six different lots of dog plasma at concentrations of 0.04,0.40 and 8.0 ng/ml were in the range of 97.6–104.9%with RSD values below 6.4%.The matrix effect of IS(1.25 ng/ml in plasma)was 93.9%and the RSD value was 6.9%.
3.3.5.Stability
Table 2 summarizes the results for the stability of melatonin, which indicated that melatonin was stable in dog plasma after storage at room temperature for 4.0 h,at?70°C for 25 d,after three freeze–thaw cycles at?70°C,after storage with dry ice for 2.0 h,and in processed samples at 4°C for 24 h.
3.4.Pharmacokinetic study and incurred sample stability
The validated LC-MS/MS method was successfully applied to pharmacokinetic study of melatonin in Beagle dogs after oral administration of 2.0 mg prolonged-release melatonin.The endogenous melatonin was not detected in all six dogs,thus it did not have impact on performing pharmacokinetic profles.The plasma concentration–time curve(mean±SD,n=6)of the analyst is shown in Fig.3.The corresponding pharmacokinetic parameters are listed in Table 3.The values of half-life andTmaxwere similar to the corresponding data reported by Yang et al.[37], although the dose employed by those authors was much higher (6.0 mg each dog).TheCmaxand AUC values of melatonin in thisstudy were dose-proportional to the published data,which was coincident with the linear-pharmacokinetics of melatonin[13]. Harpsoe et al.reported that thet1/2of melatonin was approximately 0.75 h in humans following both oral and intravenous administration routes[46].This result suggested that the elimination rate of melatonin in dogs was similar to that in humans. Yeleswaram et al.indicated that the apparent elimination halflife of melatonin following an intravenous dose of 3 mg/kg was 18.6 min in dogs[47].The signifcant shorter elimination rate might come from the different time points used for the calculation oft1/2.

Table 2–Stability of melatonin under different storage conditions(n=3).
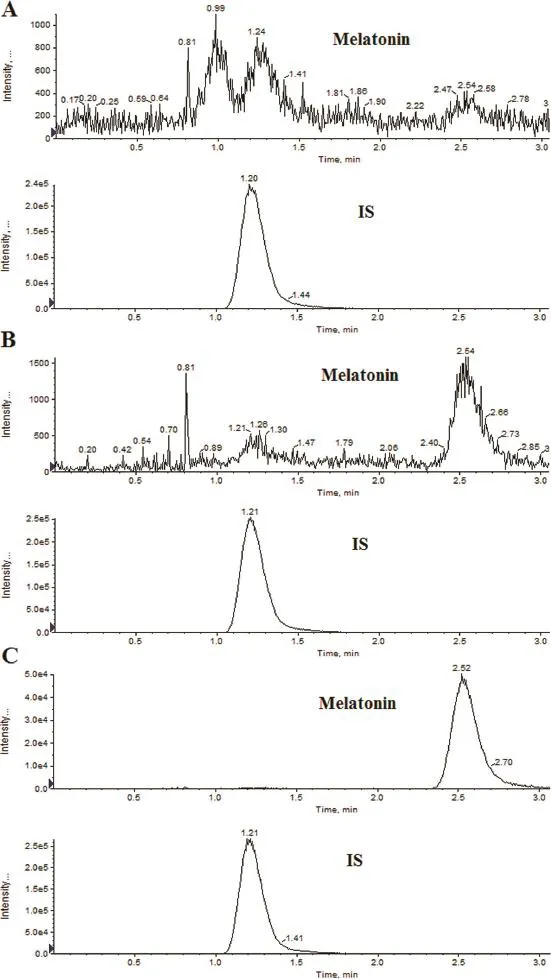
Fig.2–Representative LC-MS/MS chromatograms for melatonin and desvenlafaxine(IS).(A)Blank plasma collected at 01:00 AM on the previous day before administration;(B)blank plasma spiked with melatonin(0.020 ng/ml)and the IS;and (C)a dog plasma sample collected 2 h after oral administration of 2.0 mg prolonged-release melatonin tablets.
In the reanalysis of incurred samples,more than 92.3%of the assay satisfed the acceptance criteria.
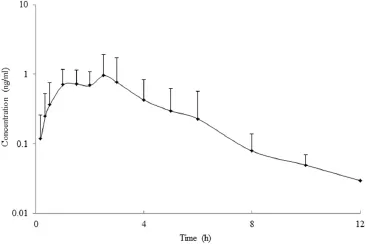
Fig.3–Mean plasma concentration–time curve of melatonin in dogs after oral administration of 2.0 mg prolonged-release melatonin tablets.The error bars are standard deviations of the mean(n=6).

Table 3–The main pharmacokinetic parameters of melatonin after oral administration of 2.0 mg prolonged-release melatonin tablets(mean±SD,n=6).
4.Conclusion
A rapid and sensitive LC-MS/MS method has been developed and validated for the analysis of melatonin in dog plasma.This method showed high throughput(3.0 min each sample)and relatively good sensitivity with an LLOQ of 0.020 ng/ml.This method was successfully applied to the pharmacokinetic study of prolonged-release melatonin in Beagle dogs.
R E F E R E N C E S
[1]Utiger RD.Melatonin–the hormone of darkness.N Engl J Med 1992;327:1377–1379.
[2]Bubenik GA,Konturek SJ.Melatonin and aging:prospects for human treatment.J Physiol Pharmacol 2011;62:13–19.
[3]Jan JE,Reiter RJ,Wasdell MB,et al.The role of the thalamus in sleep,pineal melatonin production,and circadian rhythm sleep disorders.J Pineal Res 2009;46(1):1–7.
[4]Reiter RJ,Korkmaz A.Clinical aspects of melatonin.Saudi Med J 2008;29(11):1537–1547.
[5]Zhou JN,Liu RY,Kamphorst W,et al.Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fuid melatonin levels.J Pineal Res 2003;35(2):125–130.
[6]Adi N,Mash DC,Ali Y,et al.Melatonin MT1 and MT2 receptor expression in Parkinson’s disease.Med Sci Monit 2010;16(2):BR61–BR67.
[7]Lundmark PO,Pandi-Perumal SR,Srinvasan V,et al.Role of melatonin in the eye and ocular dysfunctions.Vis Neurosci 2006;23(6):853–862.
[8]Dubocovich ML.Melatonin is a potent modulator of dopamine release in the retina.Nature 1983;306:782–784.
[9]Tamarkin L,Danforth D,Lichter A,et al.Decreased nocturnal plasma melatonin peak in patients with estrogen receptor positive breast cancer.Science 1982;216:1003–1005.
[10]Xi SC,Siu SW,Fong SW,et al.Inhibition of androgen-sensitive LNCaP prostate cancer growth in vivo by melatonin:association of antiproliferative action of the pineal hormone with MT1 receptor protein expression. Prostate 2001;46:52–61.
[11]Blask DE,Sauer LA,Dauchy RT,et al.Melatonin inhibition of cancer growth in vivo involves suppression of tumor fatty acid metabolism via melatonin receptor-mediated signal transduction events.Cancer Res 1999;59:4693–4701.
[12]Slominski A,Pisarchik A,Semak I,et al.Serotoninergic and melatoninergic systems are fully expressed in human skin. FASEB J 2002;16:896–898.
[13]DeMuro RL,Nafziger AN,Blask DE,et al.The absolute bioavailability of oral melatonin.J Clin Pharmacol 2000;40(7):781–784.
[14]Claustrat B,Brun J,Chazot G.The basic physiology and pathophysiology of melatonin.Sleep Med Rev 2005;9(1):11–24.
[15]Hardeland R.New approaches in the management of insomnia:weighing the advantages of prolonged-release melatonin and synthetic melatoninergic agonists. Neuropsychiatr Dis Treat 2009;5:341–354.
[16]Ferrua B,Masseyeff R.Immunoassay of melatonin with enzyme-labeled antibodies.J Immunoassay 1985;6:79–94.
[17]Arendt J,Paunier L,Sizonenko PC.Melatonin radioimmunoassay.J Clin Endocrinol Metab 1975;40(2):347–350.
[18]Mills MH,King MG,Keats NG,et al.Melatonin determination in human urine by high-performance liquid chromatography with fuorescence detection.J Chromatogr 1986;377:350–355.
[19]Mills MH,Finlay DC,Haddad PR.Determination of melatonin and mono-amines in rat pineal using reversed-phase ion-interaction chromatography with fuorescence detection.J Chromatogr 1991;564(1):93–102.
[20]Peniston-Bird JF,Di WL,Street CA,et al.HPLC assay of melatonin in plasma with fuorescence detection.Clin Chem 1993;39(11 Pt 1):2242–2247.
[21]Chin JR.Determination of six indolic compounds,including melatonin,in rat pineal using high-performance liquidchromatography with serial fuorimetric-electrochemical detection.J Chromatogr 1990;528(1):111–121.
[22]Vieira R,Míguez J,Lema M,et al.Pineal and plasma melatonin a determined by high-performance liquid chromatography with electrochemical detection.Anal Biochem 1992;205(2):300–305.
[23]Chanut E,Nguyen-Legros J,Versaux-Botteri C,et al. Determination of melatonin in rat pineal,plasma and retina by high-performance liquid chromatography with electrochemical detection.J Chromatogr B Analyt Technol Biomed Life Sci 1998;709(1):11–18.
[24]Musijowski J,Pobozy E,Trojanowicz M.On-line preconcentration techniques in determination of melatonin and its precursors/metabolites using micellar electrokinetic chromatography.J Chromatogr A 2006;1104:337–345.
[25]Wilson BW,Snedden W,Silman RE,et al.A gas chromatography-mass spectrometry method for the quantitative analysis of melatonin in plasma and cerebrospinal fuid.Anal Biochem 1977;81(2):283–291.
[26]Skene DJ,Leone RM,Young IM,et al.The assessment of a plasma melatonin assay using gas chromatography negative ion chemical ionization mass spectrometry.Biomed Mass Spectrom 1983;10(12):655–659.
[27]Lee CR,Esnaud H.Determination of melatonin in blood plasma,using capillary gas chromatography and electron impact medium-resolution mass spectrometry.Biomed Mass Spectrom 1988;15(5):249–252.
[28]Fourtillan JB,Gobin P,Faye B,et al.A highly sensitive assay of melatonin at the femotogram level in human plasma by gas chromatography/negative ion chemical ionization mass spectrometry.Biol Mass Spectrom 1994;23(8):499–509.
[29]Simonin G,Bru L,Lelièvre E,et al.Determination of melatonin in biological fuids in the presence of the melatonin agonist S 20098:comparison of immunological techniques and GC-MS methods.J Pharm Biomed Anal 1999;21:591–601.
[30]Wong PS,Yoshioka K,Xie F,et al.In vivo microdialysis/liquid chromatography/tandem mass spectrometry for the on-line monitoring of melatonin in rat.Rapid Commun Mass Spectrom 1999;13(5):407–411.
[31]Almeida EA,Klitzke CF,Martinez GR,et al.Synthesis of internal labeled standards of melatonin and its metabolite N1-acetyl-N2-formyl-5-methoxykynuramine for their quantifcation using an on-line liquid chromatographyelectrospray tandem mass spectrometry system.J Pineal Res 2004;36(1):64–71.
[32]Yang S,Zheng X,Xu Y,et al.Rapid determination of serum melatonin by ESI–MS–MS with direct sample injection.J Pharm Biomed Anal 2002;30:781–790.
[33]H?rtter S,Morita S,Bodin K,et al.Determination of exogenous melatonin and its 6-hydroxy metabolite in human plasma by liquid chromatography–mass spectrometry.Ther Drug Monit 2001;23:282–286.
[34]Eriksson K,Ostin A,Levin JO.Quantifcation of melatonin in human saliva by liquid chromatography-tandem mass spectrometry using stable isotope dilution.J Chromatogr B Analyt Technol Biomed Life Sci 2003;794:115–123.
[35]Wang AQ,Wei BP,Zhang Y,et al.An ultra-high sensitive bioanalytical method for plasma melatonin by liquid chromatography-tandem mass spectrometry using water as calibration matrix.J Chromatogr B Analyt Technol Biomed Life Sci 2011;879(23):2259–2264.
[36]Jensen MA,Hansen AM,Abrahamsson P,et al.Development and evaluation of a liquid chromatography tandem mass spectrometry method for simultaneous determination of salivary melatonin,cortisol and testosterone.J Chromatogr B Analyt Technol Biomed Life Sci 2011;879(25):2527–2532.
[37]Yang D,Bin D,Lin R,et al.Pharmacokinetics of melatonin sustained release tablets and common tablets in Beagle dogs.Chin J New Drugs 2011;20(23):2358–2362.
[38]Carter MD,Calcutt MW,Malow BA,et al.Quantitation of melatonin and N-acetylserotonin in human plasma by nanofow LC-MS/MS and electrospray LC-MS/MS.J Mass Spectrom 2012;47(3):277–285.
[39]Wang H,Chung-Davidson YW,Li K,et al.Quantifcation of monoamine neurotransmitters and melatonin in sea lamprey brain tissues by high performance liquid chromatography-electrospray ionization tandem mass spectrometry.Talanta 2012;89:383–390.
[40]Khan SA,George R,Charles BG,et al.Monitoring salivary melatonin concentrations in children with sleep disorders using liquid chromatography-tandem mass spectrometry. Ther Drug Monit 2013;35(3):388–395.
[41]Wang H,Walaszczyk EJ,Li K,et al.High-performance liquid chromatography with fuorescence detection and ultra-performance liquid chromatography with electrospray tandem mass spectrometry method for the determination of indoleamine neurotransmitters and their metabolites in sea lamprey plasma.Anal Chim Acta 2012;721:147–153.
[42]US Department of Health and Human Services Food and Drug Administration.Guidance for industry:bioanalytical method validation,<http://www.fda.gov/downloads/drugs/ guidancecomplianceregulatoryinformation/guidances/ ucm070107.pdf>;2001[accessed 30.04.11].
[43]Bansal S,DeStefano A.Key elements of bioanalytical method validation for small molecules.AAPS J 2007;9:E109–E114.
[44]Fast DM,Kelly M,Viswanathan CT,et al.Workshop report and follow-up–AAPS Workshop on current topics in GLP bioanalysis:assay reproducibility for incurred samples–implications of crystal city recommendations.AAPS J 2009;11(2):238–241.
[45]Uz T,Ahmed R,Akhisaroglu M,et al.Effect of fuoxetine and cocaine on the expression of clock genes in the mouse hippocampus and striatum.Neuroscience 2005;134:1309–1316.
[46]Harpsoe NG,Andersen LPH,Gogenur I,et al.Clinical pharmacokinetics of melatonin:a systematic review.Eur J Clin Pharmacol 2015;71:901–909.
[47]Yeleswaram K,McLaughlin LG,Knipe JO,et al. Pharmacokinetics and oral bioavailability of exogenous melatonin in preclinical animal models and clinical implications.J Pineal Res 1997;22:45–51.
*< class="emphasis_italic">Corresponding author.
.School of Pharmacy,Shenyang Pharmaceutical University,No.103,Wenhua Road,Shenyang 110016,China.Tel.: +86 24 23986985;fax:+86 24 23986985.
E-mail address:15142059970@163.com(X.Lu).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2015.08.004
1818-0876/?2016 The Authors.Production and hosting by Elsevier B.V.on behalf of Shenyang Pharmaceutical University.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
LC-MS/MS
Dog plasma
Pharmacokinetics
 Asian Journal of Pharmacentical Sciences2016年2期
Asian Journal of Pharmacentical Sciences2016年2期
- Asian Journal of Pharmacentical Sciences的其它文章
- Ameliorated effects of Lactobacillus delbrueckii subsp. lactis DSM 20076 and Pediococcus acidilactici NNRL B-5627 on Fumonisin B1-induced Hepatotoxicity and Nephrotoxicity in rats
- Validation and application of Vierordt’s spectrophotometric method for simultaneous estimation of tamoxifen/coenzyme Q10 in their binary mixture and pharmaceutical dosage forms
- Introduction of antineoplastic drug NSC631570 in an inpatient and outpatient setting: Comparative evaluation of biological effects
- Control of autoimmune arthritis by herbal extracts and their bioactive components
- The 1st Euro-Mediterranean Workshop:Natural Products in Health and Diseases:Cairo,Egypt, March 2,2015
- Effect of process parameters on the recrystallization and size control of puerarin using the supercritical fuid antisolvent process