
Therapeutic resistance in cancer: microRNA regulation of EGFR signaling networks
2013-09-26 06:01:14GermanGomezJillWykoskyCiroZancaFrankFurnariWebsterCavenee
Cancer Biology & Medicine 2013年4期
German G.Gomez, Jill Wykosky, Ciro Zanca, Frank B.Furnari, Webster K.Cavenee
Ludwig Institute for Cancer Research, University of California San Diego, La Jolla, CA 92093, USA
Introduction
The human ErbB/epidermal growth factor receptor (EGFR)family is comprised of four members (EGFR/ErbB1, ErbB2,ErbB3 and ErbB4) that transduce signals upon binding to ligands to regulate important cellular processes such as cell division,differentiation, migration and programmed cell death1.Ligand binding induces EGFR dimerization, transphosphorylation of dimerized receptors and ultimately tyrosine kinase activation.Activated EGF-receptors recruit multiple adaptor and effector proteins and then initiate signaling through the PI3K, MAPK and STAT3 pathways to regulate a multitude of cellular activities1,2.
Since EGFR regulates fundamental cellular processes, it is not surprising that misregulation of EGFR signaling occurs frequently in several types of tumors including glioblastoma(GBM), colorectal cancer (CRC), head and neck squamous cell carcinoma (HNSCC), non-small cell lung cancer (NSCLC),breast, renal, ovarian, bladder, prostate and pancreatic cancers3-5.Consequently, multiple therapeutic agents and strategies have been developed to block the strong tumor promoting effects exerted by EGFR6.While some patients have shown encouraging responses to anti-EGFR therapies, durable responses are uncommon2.A thorough understanding of the factors that dictate response to EGFR inhibitors might spark the design of novel therapeutics to combat the development of resistance to such inhibitors.
MicroRNAs (miRs) are a novel group of non-coding small regulatory RNAs, which finely tune gene expression7and are emerging as unique effector molecules of the different signaling cascades initiated by EGFR in normal and transformed cells.Here, we emphasize the role of the miRs most commonly involved in facilitating or suppressing aberrant EGFR signaling in a variety of tumor types.Additionally, we highlight the possibility of using miRs or anti-miR oligonucleotides as novel therapeutic agents to overcome resistance to anti-EGFR therapies.We also present evidence supporting the use of miRs and their targets as molecular predictors of response to EGFR inhibitors.
Mechanisms of aberrant EGFR activation in cancer
Aberrant EGFR signaling occurs through a variety of mechanisms, including overexpression as a consequence of gene amplification, genetic mutations, cross-talk between mutant and wild-type EGFR, excessive levels of activating ligands or autocrine signaling and altered EGFR cellular localization.
EGFR overexpression and gene amplification
Under normal physiological conditions, EGFR is present at about 4×104to 1×105EGFR molecules/cell8,9.In contrast, tumors can express about 5×105to 2×106EGFR molecules/cell10-12.In GBM,the EGFR gene is amplified to very high levels (>20 copies/cell)13and in about 50% of primary GBMs where it is associated with poor prognosis14-18, as compared to secondary GBM patients19.
Mutations
Various EGFR mutations are well-documented and shown to be tumor-type specific.One mutation with profound pathologic effects is a truncated form of EGFR, commonly found in about 50% of GBMs with EGFR amplification9, named EGFRvIII(also known as ΔEGFR, EGFR* or de2-7EGFR)20.EGFRvIII lacks a portion of the extracellular ligand-binding domain, is constitutively active and slowly recycled.EGFRvIII does not bind ligand but initiates constitutive mitogenic and cell survival signals21, and a resulting worse prognosis for GBM patient whose tumors express it22.Other truncation mutants of EGFR(EGFRvII; EGFRvV) have less demonstrated clinical relevance23(Figure 1).
Point mutations in the intracellular portion of the EGFR gene have been found in different tumor types, in particular in the EGFR kinase domain in NSCLC (Figure 1)24,25.The L858R mutation and deletions in exon 19 confer constitutive kinase activity and are associated with a better response of lung tumors to EGFR kinase inhibitors26-28.
Tumor microenvironment
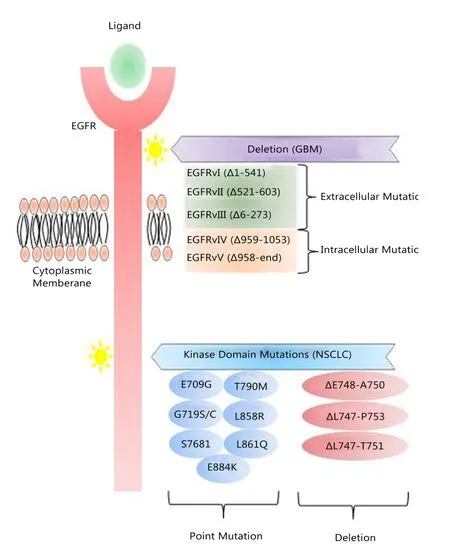
Figure 1 EGFR mutations in GBM and NSCLC.Tumor-type specific mutations of EGFR are well-established.In GBM, EGFR undergoes deletion processes that lead to the expression of truncated mutants,of which EGFRvIII is the major form that is associated with a poor response to conventional and EGFR-TKIs therapies.Point mutations of the EGFR kinase domain are predominant in NSCLC.
Despite the strong tumorigenic effects exerted by EGFRvIII,EGFRvIII-expressing cells are not the predominant subpopulation in a brain tumor cell mass, which is often represented by wild-type EGFR expressing cells29.This phenomenon raised the possibility that EGFRvIII drives tumorigenesis in cooperation with wild-type EGFR.In a model of GBM heterogeneity, EGFRvIII-expressing cells produce secreted factors, such as IL-6 and LIF, which leads to cytokine co-receptor gp130-wild-type EGFR cross-talk and subsequent transactivation of wild-type EGFR to promote tumorigenesis30.Interestingly, hypoxia has been proposed to enhance EGFR-mRNA translation, thus providing another mechanism of increased EGFR expression and signaling31.
Ligand-mediated activation
EGFR contains four extracellular domains (DI, DII, DIII and DIV), among which DI and DIII are required for ligand binding32.Ligand binding causes EGFR to form homodimers, or heterodimers with other ErbB family members (ErbB2, ErbB3,ErbB4), thus leading to tyrosine kinase domain activation and signal transduction5.Under physiological conditions, many ligands bind and activate EGFR, including epidermal growth factor, transforming growth factor-alpha, heparin-binding EGF,betacellulin, epigen, epiregulin, amphiregulin and neuregulin 2b33.Different ligands might have different downstream effects,but the mechanism remains unclear34.Ligand overexpression in cancer leads to persistent EGFR signaling35,36.
Cellular localization
Membrane-associated EGFR signaling is augmented by heterodimerization with other members of the EGFR family,such as ErbB237.Importantly, EGFR signaling is not completely restricted to the plasma membrane.EGFR localizes to the nucleus of cancer cells and its nuclear localization is associated with a poor prognosis in different types of cancers38-45.Nuclear EGFR appears to promote cell proliferation through its tyrosine kinase activity or by acting as a transcriptional regulator46,47.EGFR has also been found in the mitochondria48, thus providing a new perspective on EGFR subcellular location and its role in cancer.
EGFR-targeted therapies
As EGFR promotes oncogenesis by activating the signaling pathways that regulate tumor formation and progression, it is not surprising that it has become one of the most heavily targeted molecules for therapy.The three most common agents for targeting EGFR are monoclonal antibodies (mAbs), vaccines,and small molecule inhibitors.
Antibodies
The mAbs cetuximab49-51and fully humanized panitumumab52,53are specific for the extracellular domain of EGFR and display anti-tumor activity in patients.These antibodies, which are FDA-approved for use in the U.S., bind to EGFR and prevent ligand-mediated activation of the receptor.Another antibody,mAb 806, binds not only to amplified wild-type EGFR but also more strongly to the mutant EGFRvIII54,55.This antibody is advantageous because the mutant receptor is not expressed in normal tissue, so should minimize side effects resulting from antibody binding to EGFR expressed on non-tumor cells.While the primary anti-tumor activity of EGFR mAbs is attributed to blocking receptor activation, other mechanisms likely contribute to the anti-tumor effects such as receptor downregulation56and antibody-dependent cellular cytotoxicity (ADCC)57.
Anti-EGFR vaccines
Another approach for targeting EGFR is vaccines that elicit an immune response against EGFR-expressing tumor cells.One vehicle is dendritic cells pulsed with EGFR-specific antigenic peptides58.CDX-110 is a peptide vaccine that induces anti-tumor immune responses to EGFRvIII positive cells; this vaccine has shown pre-clinical efficacy and some encouraging clinical results59.An alternate approach which is becoming increasingly more common is the engineering of T lymphocytes to express chimeric antigen receptors (CARs).EGFR-targeted CAR T cells have demonstrated anti-tumor efficacy both in vitro and in vivo with low systemic toxicity60.
Small molecules
Small molecule inhibitors that compete with ATP for binding to the active conformation of the EGFR kinase domain are perhaps the most widespread approach to targeting this receptor61.The orally bioavailable reversible inhibitor, erlotinib,is FDA-approved for the treatment of NSCLC, and its closely related cousin, gefitinib, is approved for multiple solid tumors in countries outside the U.S.Afatinib is a second-generation,irreversible EGFR/ErbB2 inhibitor that has recently gained FDA approval for EGFR-mutant NSCLC along with a companion diagnostic test to determine the EGFR mutation status62,63.
Mechanisms of resistance to EGFR inhibitors
While these targeted therapeutic approaches are rational strategies, clinical bene fit from them is rare.Response is typically observed in only a subset of patients, especially in an unselected patient population.Thus, the decision to begin and continue treatment must be based upon reliable biomarkers that are predictive of response.Specific mutations in the kinase domain of EGFR render some NSCLC tumors exquisitely dependent on EGFR-mediated signaling for survival and are predictive of response to EGFR-tyrosine kinase inhibitors (TKIs)26-28.In fact,the presence of these mutations in NSCLC tumor biopsy tissue is now required in order for a patient to receive TKI therapy.In nearly all other tumor types, however, there does not appear to be a specific EGFR mutation that predicts for response to these therapies, and it has become clear that expression of the target alone does not suffice as a predictive biomarker64.Rather, it appears that more complex mechanisms underlie the response of tumors to EGFR-targeted therapy.
Resistance to EGFR-targeted therapy following an initial clinical response to these drugs is a common clinical observation.Although mechanisms, such as the presence of mutations in codons 12 and 13 of the K-Ras gene65exist that render tumor cells inherently resistant to EGFR-targeted therapies, acquired resistance is a more widespread clinical problem.Thus, the mechanisms of and methods to overcome resistance are an area of intense study.In the case of EGFR-targeted antibodies and vaccines, resistance almost always manifests as the outgrowth of a population of tumor cells that are devoid of EGFR expression66meaning that they have “escaped” the targeted therapy by eradicating the target.
T790M EGFR mutation
Treatment of NSCLC with the EGFR-TKIs gefitinib and erlotinib results in the generation of a second-site mutation in the EGFR kinase domain that changes a threonine to a methionine—T790M—in at least 50% of patients67.This mutation increases the affinity of the kinase domain for ATP while decreasing affinity for the small molecules and the consequent decrease of drug binding causes sustained phosphorylation of EGFR and clinical drug resistance.One approach to circumvent T790M-mediated resistance has been the use of irreversible small molecule inhibitors such as afatinib, which has demonstrated activity against T790M mutant cells and tumors in preclinical models68.Disappointingly, this has resulted in only a modest effect in patients who progressed during prior treatment with erlotinib or ge fitinib69.
Alternative receptor tyrosine kinase activation
While the T790M mutation represents a direct mechanism for drug resistance that occurs at the level of drug accessibility to the target, there are a number of indirect mechanisms that involve the increased expression or activation of alternative growth factor receptors to maintain signal flux, despite EGFR inhibition.Perhaps the best characterized of these is the amplification of the Met receptor70and/or elevated levels of its ligand HGF71.In gefitinib resistant NSCLC cell lines, Met drives ErbB3-dependent activation of the PI3K pathway72.In addition, increased expression and activity of Met and ErbB3 are associated with resistance to cetuximab73.The receptor Axl was also identified as a potential target for overcoming EGFR inhibitor resistance associated with epithelial to mesenchymal transition74.Interestingly, HGF has recently been shown to drive resistance through stimulation of EGFR binding to Axl and the EphA2 receptor in a kinase-independent fashion75.
PI3K activation and PTEN inactivation
PI3K signaling pathway activation is a frequently observed mechanism of resistance.One way to achieve elevated levels of PI3K signaling and resistance to targeted EGFR inhibition is by direct mutation of PIK3CA, the gene encoding the p110 catalytic and p85 regulatory subunits of PI3K.In colorectal tumors,PIK3CA mutations are associated with reduced sensitivity to cetuximab76.In GBM, patients who responded to EGFRTKIs demonstrated co-expression of the mutant EGFRvIII and PTEN, a negative regulator of PI3K activity77.It appears that for EGFR targeting to be effective, complete inhibition of PI3K activity must be achieved.Tumors lacking PTEN expression generally have elevated and sustained PI3K pathway activity,and restoration of functional PTEN re-sensitizes resistant cells to erlotinib78.Furthermore, EGFR-TKI resistance is associated in some cases with FGFR and Src family kinase-mediated phosphorylation of PTEN at tyrosine 24079.Similar persistent signaling can be achieved by the co-activation of other RTKs such as PDGFR80.
Some of the scenarios mentioned above point toward rational strategies for targeting that take advantage of new therapeutic vulnerabilities arising as a result of the specific resistance mechanism.Combination therapies or salvage therapies utilizing other RTK inhibitors or specific inhibitors to pathway components that contribute to resistance are being explored, both pre-clinically and clinically.One striking example is the induction of the promyelocytic leukemia (PML) gene following EGFR-TKI-mediated inhibition of mTOR signaling81.Expression of PML confers sensitivity to arsenic trioxide and points toward a novel therapeutic strategy for resistant tumors with elevated PML.
MicroRNAs regulate EGFR signaling and susceptibility to EGFR inhibitors
MicroRNAs
MicroRNAs (miRs) are a group of non-protein encoding RNAs that are 19-25 nt in length and block translation or facilitate mRNA degradation upon binding to complementary sequences in the 3' UTR of their target mRNAs7.The first miR, lin-4, was discovered 20 years ago where it was shown to decrease lin-14 protein expression in C.elegans82.Subsequent studies identified the existence of new miRs in several species including mice and humans83-85and approximately 900 miRs have been so far identified86.
MiRs are transcribed by RNA polymerase II as large primary transcripts (pri-miRs) that are processed by Drosha/DGCR8 complexes to yield 60-110 nt long hairpin containing-precursor miRs (pre-miRs)87.After export of the pre-miRs to the cytoplasm by exportin-588, mature miRs are excised from the pre-miRs by the Rnase III enzyme, Dicer89, and loaded into the RNA-induced silencing complex (RISC)90.There, mature miRs are guided to their appropriate target mRNAs to prevent translation.A recently discovered alternate and conserved miR biogenesis pathway,the miRtron pathway, generates pre-miRs through splicing mechanisms that do not require Drosha/DGCR8 activity91,92.
Ample evidence indicates that the de-regulation of miR expression and activity, as a consequence of genomic alterations93, miR gene methylation94, aberrant transcription95and defective miR processing96,97, are intimately involved in cancer initiation, maintenance, and progression7.Below we focus on the regulation of EGFR signal networks by miRs in cancer and the involvement of miRs in facilitating resistance to EGFR-inhibition (Table 1).
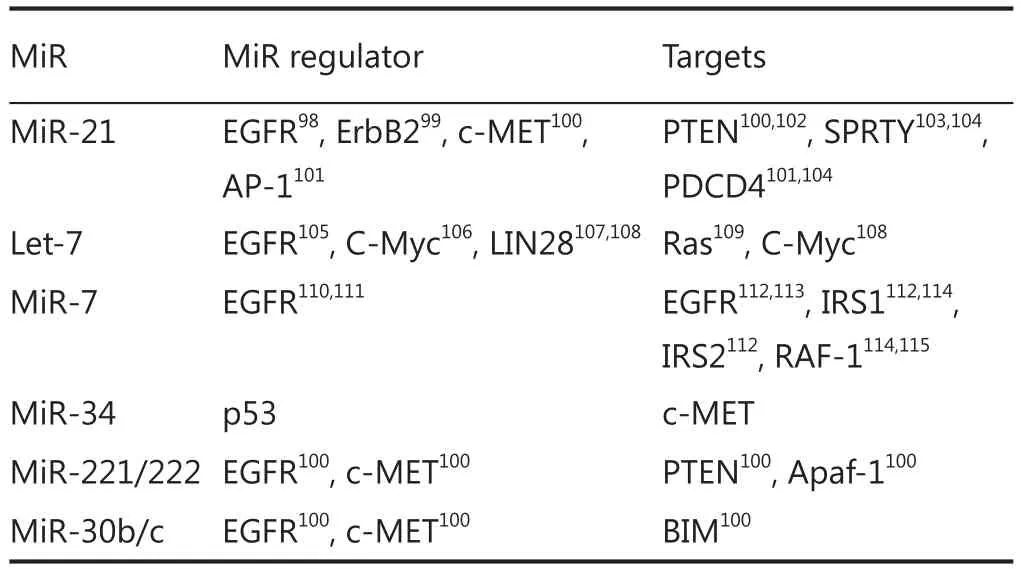
Table 1 MicroRNAs that sustain or repress EGFR signaling
MiR-21
MiR-21 is a bona-fide “oncomir” and one of the most widely studied miRs due to its dramatic upregulation in many cancers,ability to target the tumor suppressor PTEN and thereby reducing tumor susceptibility to TKIs77, as well as its regulated expression in hypertrophic heart disease models116.Significant overexpression of miR-21 was observed in primary GBM specimens and cultured cells relative to normal brain tissues and cells.Moreover, functional inhibition of miR-21activity using 2′-O-methyl and locked nucleic acid oligonucleotides revealed miR-21 to be an anti-apoptotic miR117.Subsequent studies have focused on clarifying the mechanisms of miR-21 deregulation in cancer, identifying miR-21 targets and their involvement in therapeutic response and the potential of miR-21 to serve as a novel cancer target.
In GBM, inhibition of miR-21 activity increased PTEN expression and decreased tumorigenicity, EGFR expression and Akt activation102.MiR-21 is positively regulated by EGFR in cancer cells as demonstrated by the finding that AG1478, an EGFR-TKI, blocked EGFR induction of miR-2198.Interestingly,activation of the EGFR family member, ErbB2, induced miR-21 to promote cell invasion through suppression of the well-established miR-21 target, PDCD499.Critically, oncogenic HRasG12Vwas sufficient to induce miR-2199, consistent with oncogenic Ras mutants conferring resistance to EGFR-TKIs65.The discovery of a novel autoregulatory loop revealed that miR-21 targets PDCD4, a negative AP-1 regulator, upon its induction by AP-1 in response to Ras signaling101, and several other studies con firmed that miR-21 is positively regulated by Ras/ERK signaling103,118,119.Interestingly,data from transgenic tumor models show that miR-21 drives tumorigenesis by repressing negative regulators of the Ras/MEK/ERK, Ras/PI3K/Akt and Ras/RalGDS/JNK pathways, thus further demonstrating that miR-21 acts as an effector of Ras to promote transformation103,104.Collectively, these reports suggest that EGFR family members positively regulate miR-21 as a means to achieve a signaling threshold required for transformation and the maintenance of malignant cellular phenotypes (Figure 2).

Figure 2 EGFR induces miR-21 to inhibit negative regulators of downstream EGFR pathways.Induction of miR-21 occurs upon AP-1 activation in response to EGFR/RalGDS/ JNK signaling.MiR-21 targets the PDCD4 and PTEN tumor suppressors to achieve maximal Ras/RalGDS/JNK/AP-1 and Ras/PI3K/Akt signaling.MiR-21 targets the ligand-induced negative RTK feed-back regulator, SPRY, to maintain prolonged Ras/Raf/Erk signaling.
The ability of miR-21 to inhibit apoptosis and sustain the activation of oncogenic signaling pathways led to the proposal and subsequent demonstration that its expression could predict and modify responses to conventional cancer therapies120,121.With regard to resistance to EGFR-TKIs, miR-21 blockade is able to reverse the EMT phenotype associated with EGFR-TKI resistance74,122,123.In human breast cancer models miR-21 upregulation caused acquired resistance to the anti-HER2/neu antibody, Trastuzumab124.Trastuzumab-resistant cells showed decreased PTEN expression that was restored upon inactivation of miR-21.When combined with Trastuzumab, miR-21 antisense oligonucleotide therapy significantly inhibited the growth of Trastuzumab-resistant tumors.Importantly, miR-21 overexpression was correlated with reduced PTEN levels in Trastuzumab-resistant breast cancer patients124.Gefitinib-resistant lung cancer cells having high levels of miR-21, miR-30b/c and miR-221/22, as consequence of c-MET overexpression, were rendered susceptible to gefitinib when treated with SU11274, a c-MET inhibitor100.Combined EGFR and c-MET inhibition induced PTEN, Apaf-1 and BIM, as a consequence of downregulation of miR-21, miR-30b/c and miR-221/221100.Collectively, these reports suggest that EGFR and c-Met coordinately regulate multiple miRs to finetune downstream signaling cascades that render tumors resistant to EGFR-TKIs (Figure 3).
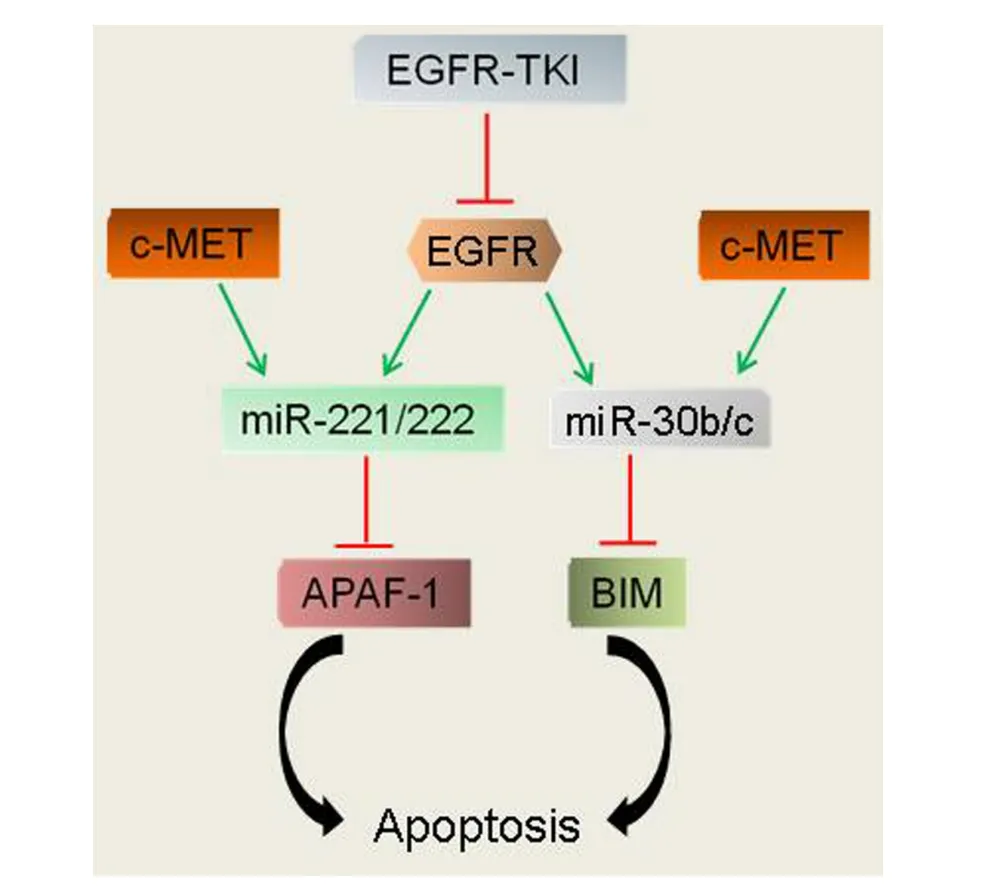
Figure 3 A model of resistance to EGFR-TKIs.EGFR induction of the miR-221/222 and miR-30b/c family members represses the proapoptotic genes Apaf-1 and BIM, rendering cells less susceptible to apoptotic cell death.EGFR-TKIs reverse the suppression of Apaf-1 and BIM to induce apoptosis of EGFR-dependent tumor cells.Amplication and/or activation of c-MET restores the induction of miR-221/222 and miR-30b/c and, consequently, the repression of Apaf-1 and BIM to escape EGFR-TKI-mediated apoptosis.
Let-7
The let-7 family members are highly conserved and reside in regions of the human genome frequently altered in cancers93,125.Let-7 was first shown to behave as a tumor suppressor in a study where it inhibited lung cancer growth126.In addition, the observed downregulation of let-7 family members correlated with shorter survival in a cohort of 143 lung cancer cases.The mechanism of tumor suppression by let-7 was revealed when HRas, NRas and KRas were shown to be let-7 targets109and that let-7 suppression was necessary for tumor initiation,maintenance and metastasis114,127.
The frequent suppression of let-7 in cancers prompted studies to identify the mechanisms regulating its stability and biogenesis.The demonstration that the LIN28 RNA binding protein selectively impairs processing of the let-7 family led to the verification that inhibition of let-7 processing by LIN28 and LIN28B is sufficient to transform cells107,108.Furthermore, LIN28 is overexpressed in a subset of human cancers where it induces expression of two let-7 targets, KRas and c-Myc, to transform NIH3T3 cells.Interestingly, c-Myc positively regulates LIN28B to repress let-7 family members106, suggesting the possibility that an RTK/Ras/ERK/C-Myc signaling loop is ultimately responsible for let-7 inactivation.Supporting this hypothesis,SHP2, a protein tyrosine phosphatase required for maximal ERK activation downstream of grow factor receptors, activated c-Myc, and this resulted in the repression of let-7 and promotion of breast cancer maintenance128.These interactions were underscored by finding that inhibition of EGFR kinase activity with gefitinib induced let-7c, showing that EGFR signaling suppresses let-7105.
Mutant forms of the Ras proteins occur in certain tumors where they confer protection to cytotoxic therapies65.Experimental manipulation of the LIN28-let-7-KRas regulatory network by let-7 overexpression and LIN28 silencing caused radio-sensitization of KRas mutant lung and pancreatic cancer cells129, suggesting that let-7 and KRas might serve as markers of survival and therapeutic response.In patients with KRasmutated mCRC, high let-7a expression was predictive of better overall and progression-free survival130.Interestingly, while low let-7 expression and KRas mutation expectedly predict poor survival126,131, a variant KRas, KRas-LCS6, is associated with increased risk of developing NSCLC and reduced survival in HNSCC132,133.The KRas-LCS6 allele bears a single nucleotide polymorphism (SNP), is present in about 20% of NSCLC tumors analyzed and disrupts let-7 binding to the 3’UTR of KRas.DNA sequencing of KRas in mCRC tissues obtained from patients that had undergone salvage irinotecan-cetuximab therapy showed that KRas-LCS6 positive tumors responded poorly to the therapy relative to tumors without the LCS6 SNP134.These initial observations were supported by other results that linked the KRas-LCS6 with non-response to anti-EGFR therapy in tumors bearing wild type KRas and BRAF135.Collectively, these reports suggest that the disruption of the let-7-KRas regulatory network by the KRas-LCS6 SNP is predictive of response to anti-EGFR therapy in CRCs with wild type KRas and BRAF.
MiR-7
MiR-7 was first implicated as an effector of EGFR signaling in studies of Drosophila where inactivation of the Yan transcription factor through the EGFR/ERK signaling axis induces miR-7 to promote photoreceptor differentiation110.In NSCLC,miR-7 induction by both wild type and mutant EGFR L858R required the RAS/ERK/c-Myc signaling axis111to promote lung tumorigenesis by repressing the transcriptional regulator ERF.Supporting the oncogenic role of miR-7, molecular diagnostic testing showed that 60% of NSCLC fine-needle aspirates had upregulation of miR-7136.MiR-7 was also significantly upregulated in renal cell carcinoma (RCC) samples relative to normal tissues and miR-7 was required to promote RCC survival, proliferation and migration137.
Paradoxically, several studies have shown that miR-7 also acts as tumor suppressor by directly targeting EGFR itself.However,the categorization of miRs as tumor suppressors or oncogenes must take into account the cellular context as this dictates the functions of miRs138.It was first demonstrated in GBM that miR-7 targets EGFR and that impairment of miR-7 processing leads to its downregulation112.In addition, miR-7 inhibited GBM cell proliferation, survival and migration while also inhibiting Akt signaling by targeting insulin receptor substrates, IRS-1 and IRS-2.MiR-7 also attenuates the activation of Akt and ERK that is induced by EGFR signaling in multiple cancer cell types113.In addition to downregulating EGFR, miR-7 targets several other genes involved in EGFR signaling and tumorigenesis, indicating that miR-7 negatively regulates EGFR signaling in several types of cancers.Subsequent studies confirmed that miR-7 inhibits EGFR, and its downstream signaling components, to negatively regulate tumor cell migration, invasion, metastasis and tumorigenesis in various tumor cell types139-141.Collectively,these observations provided the impetus to target EGFR signaling networks using miR-7 to circumvent resistance to conventional and targeted therapies.
In vitro radiosensitivity experiments were employed to determine the ability of miR-7 to reverse the radioresistance conferred on human cancer cells by EGFR142.As would be predicted from the prior studies cited above, miR-7 blunted EGFR/PI3K/Akt signaling and reversed the radio-resistance.Correspondingly, direct injection of a liposome-encapsulated miR-7 plasmid into established EGFR-TKI sensitive and resistant tumors bearing the T790M EGFR mutant resulted in significant tumor regression in conjunction with repression of EGFR, RAF-1 and IRS-1 expression142.In head and neck cancer,miR-7 functioned in a synergistic manner with erlotinib to render FaDu Erlotinib-resistant cells susceptible to the growth inhibitory activities of the drug115.Interestingly, expression profiling analysis suggested that the downregulation of RAF-1 and EGFR and its ligand TGF-α by miR-7 was a possible mechanism by which miR-7 orchestrates the inhibition of EGFR signaling at multiple levels115.In total, these observations show promise for miR-7-based strategies to effectively target EGFR addicted and EGFR-TKI resistant tumors.
MiR-34
The p53 tumor suppressor senses DNA damage, cellular stress and inappropriate mitogenic cues.In response to such signals,p53 facilitates DNA repair, induces cell death and arrests cell division143.The p53 pathway requires the induction and activation of many gene products to regulate a diverse array of cellular stress responses.In 2007, it was shown that p53 induces expression of members of the miR-34 family in response to ionizing radiation144.The regulation of miR-34 family members by p53 and their involvement in p53-induced cell death and cell cycle arrest was then validated in other model systems145-147.
Of relevance to EGFR-TKI resistance, as c-Met overexpression confers resistance to EGFR-TKIs70, miR-34 directly targeted cell cycle-related proteins and c-Met in MEF cells144.In GBM and ovarian cancer, miR-34a/b/c expression was inversely correlated with c-Met expression148,149.In addition,miR-34 inhibited cell invasion, proliferation and tumorigenesis,while c-Met overexpression partially reversed the cell death and cell cycle arrest induced by miR-34149,150.C-Met was established as a bona fide miR-34 target in melanoma, lung, colon, breast and gastric cancer cells151.Importantly, miR-34 inhibited activation of c-Met, Akt, ERK and it impaired c-Met driven invasion.
Given c-Met’s ability to promote tumor cell motility,invasion and resistance to EGFR-TKIs, c-Met inhibition has been hypothesized to limit tumor spreading and resistance to cytotoxic agents and targeted therapies152.In support, the induction of miR-34 by p53 downregulated c-Met and inhibited c-Met mediated tumor cell motility and invasion153.Combined treatment of hepatocellular carcinoma cells with miR-34a and the c-Met inhibitor, SU11274, significantly induced cell death and inhibited cell proliferation154.To explore the feasibility of miR-34a as an anti-tumor agent, the ability of miR-34 lenti-virus to suppress the growth of therapeutically resistant lung cancers bearing KRas and p53 mutations was determined155.As anticipated, KRas/p53 mutant tumors showed decreased miR-34 expression and increased c-Met levels relative to normal lung and lenti-viral delivery of miR-34 impaired lung tumor initiation and progression.These findings suggest that miR-34 replacement therapies might sensitize resistant tumors to EGFR-TKIs by suppressing c-Met expression and its activation of oncogenic signaling pathways.
Conclusion
There is a rapidly increasing understanding of EGFR signaling,therapeutic targeting and mechanisms of resistance.There is also the accumulation of a large body of knowledge about miRs and their intimate involvement in tumorigenesis and tumor progression.This has led to the recognition of mechanisms by which tumors modulate miR activities to thrive when subjected to selective pressures applied by therapy.It is evident that miRs represent a novel group of regulatory RNAs that EGFR impinges upon to promote its tumorigenic activities.The ability to modulate miR activity and/or the activity of miR targets suggests a novel therapeutic avenue to overcome resistance mechanisms to conventional and EGFR-targeted therapies.
Acknowledgements
This work was supported by An American Brain Tumor Association Basic Research grant to G.G.G.in memory of Keith Powers, P01-CA95616, R01-NS080939 and James S.McDonnell Foundation.
Conflict of interest statement
No potential conflicts of interest are disclosed.
1.Huang PH, Xu AM, White FM.Oncogenic EGFR signaling networks in glioma.Sci Signal 2009;2: re6.
2.Taylor TE, Furnari FB, Cavenee WK.Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance.Curr Cancer Drug Targets 2012;12:197-209.
3.Wheeler DL, Dunn EF, Harari PM.Understanding resistance to EGFR inhibitors-impact on future treatment strategies.Nat Rev Clin Oncol 2010;7:493-507.
4.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al.Emerging insights into the molecular and cellular basis of glioblastoma.Genes Dev 2012;26:756-784.
5.Yewale C, Baradia D, Vhora I, Patil S, Misra A.Epidermal growth factor receptor targeting in cancer: A review of trends and strategies.Biomaterials 2013;34:8690-8707.
6.Wykosky J, Fenton T, Furnari F, Cavenee WK.Therapeutic targeting of epidermal growth factor receptor in human cancer:successes and limitations.Chin J Cancer 2011;30:5-12.
7.Calin GA, Croce CM.MicroRNA signatures in human cancers.Nat Rev Cancer 2006;6:857-866.
8.Carpenter G, Cohen S.Epidermal growth factor.Annu Rev Biochem 1979;48:193-216.
9.Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN, Cavenee WK et al.A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity.Proc Natl Acad Sci U S A 1994;91:7727-7731.
10.Petrides PE, Bock S, Bovens J, Hofmann R, Jakse G.Modulation of pro-epidermal growth factor, pro-transforming growth factor alpha and epidermal growth factor receptor gene expression in human renal carcinomas.Cancer Res 1990;50:3934-3939.
11.Yao M, Shuin T, Misaki H, Kubota Y.Enhanced expression of c-myc and epidermal growth factor receptor (C-erbB-1) genes in primary human renal cancer.Cancer Res 1988;48:6753-6757.
12.Wikstrand CJ, McLendon RE, Friedman AH, Bigner DD.Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII.Cancer Res 1997;57:4130-4140.
13.Lee JC, Vivanco I, Beroukhim R, Huang JH, Feng WL, DeBiasi RM, et al.Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain.PLoS Med 2006;3:e485.
14.Hurtt MR, Moossy J, Donovan-Peluso M, Locker J.Amplification of epidermal growth factor receptor gene in gliomas:histopathology and prognosis.J Neuropathol Exp Neurol 1992;51:84-90.
15.Jaros E, Perry RH, Adam L, Kelly PJ, Crawford PJ, Kalbag RM,et al.Prognostic implications of p53 protein, epidermal growth factor receptor, and Ki-67 labelling in brain tumours.Br J Cancer 1992;66:373-385.
16.Lenz HJ, Van Cutsem E, Khambata-Ford S, Mayer RJ, Gold P, Stella P, et al.Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines.J Clin Oncol 2006;24:4914-4921.
17.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al.KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer.Cancer Res 2006;66:3992-3995.
18.Schlegel J, Stumm G, Br?ndle K, Merdes A, Mechtersheimer G, Hynes NE, et al.Amplification and differential expression of members of the erbB-gene family in human glioblastoma.J Neurooncol 1994;22:201-207.
19.Watanabe K, Tachibana O, Sata K, Yonekawa Y, Kleihues P, Ohgaki H.Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas.Brain Pathol 1996;6:217-223; discussion 23-24.
20.Gan HK, Kaye AH, Luwor RB.The EGFRvIII variant in glioblastoma multiforme.J Clin Neurosci 2009;16:748-754.
21.Nagane M, Levitzki A, Gazit A, Cavenee WK, Huang HJ.Drug resistance of human glioblastoma cells conferred by a tumorspecific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases.Proc Natl Acad Sci U S A 1998;95:5724-5729.
22.Jones TS, Holland EC.Molecular pathogenesis of malignant glial tumors.Toxicol Pathol 2011;39:158-166.
23.Frederick L, Wang XY, Eley G, James CD.Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas.Cancer Res 2000;60:1383-1387.
24.J?nne PA, Engelman JA, Johnson BE.Epidermal growth factor receptor mutations in non-small-cell lung cancer: implications for treatment and tumor biology.J Clin Oncol 2005;23:3227-3234.
25.Sharma SV, Bell DW, Settleman J, Haber DA.Epidermal growth factor receptor mutations in lung cancer.Nat Rev Cancer 2007;7:169-181.
26.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA,Brannigan BW, et al.Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to ge fitinib.N Engl J Med 2004;350:2129-2139.
27.Paez JG, J?nne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al.EGFR mutations in lung cancer: correlation with clinical response to ge fitinib therapy.Science 2004;304:1497-1500.
28.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al.EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to ge fitinib and erlotinib.Proc Natl Acad Sci U S A 2004;101:13306-13311.
29.Nishikawa R, Sugiyama T, Narita Y, Furnari F, Cavenee WK,Matsutani M.Immunohistochemical analysis of the mutant epidermal growth factor, deltaEGFR, in glioblastoma.Brain Tumor Pathol 2004;21:53-56.
30.Inda MM, Bonavia R, Mukasa A, Narita Y, Sah DW, Vandenberg S, et al.Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma.Genes Dev 2010;24:1731-1745.
31.Franovic A, Gunaratnam L, Smith K, Robert I, Patten D, Lee S.Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer.Proc Natl Acad Sci U S A 2007;104:13092-13097.
32.Lax I, Fischer R, Ng C, Segre J, Ullrich A, Givol D, et al.Noncontiguous regions in the extracellular domain of EGF receptor define ligand-binding specificity.Cell Regul 1991;2:337-345.
33.Yarden Y.The EGFR family and its ligands in human cancer.signalling mechanisms and therapeutic opportunities.Eur J Cancer 2001;37 Suppl 4:S3-8.
34.Wong L, Deb TB, Thompson SA, Wells A, Johnson GR.A differential requirement for the COOH-terminal region of the epidermal growth factor (EGF) receptor in amphiregulin and EGF mitogenic signaling.J Biol Chem 1999;274:8900-8909.
35.Rusch V, Klimstra D, Venkatraman E, Pisters PW, Langenfeld J,Dmitrovsky E.Overexpression of the epidermal growth factor receptor and its ligand transforming growth factor alpha is frequent in resectable non-small cell lung cancer but does not predict tumor progression.Clin Cancer Res 1997;3:515-522.
36.Vermi W, Giurisato E, Lonardi S, Balzarini P, Rossi E, Medicina D, et al.Ligand-Dependent Activation of EGFR in Follicular Dendritic Cells Sarcoma is Sustained by Local Production of Cognate Ligands.Clin Cancer Res 2013;19:5027-5038.
37.Erjala K, Sundvall M, Junttila TT, Zhang N, Savisalo M, Mali P,et al.Signaling via ErbB2 and ErbB3 associates with resistance and epidermal growth factor receptor (EGFR) amplification with sensitivity to EGFR inhibitor ge fitinib in head and neck squamous cell carcinoma cells.Clin Cancer Res 2006;12:4103-4111.
38.Cao H, Lei ZM, Bian L, Rao CV.Functional nuclear epidermal growth factor receptors in human choriocarcinoma JEG-3 cells and normal human placenta.Endocrinology 1995;136:3163-3172.
39.Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, et al.Nuclear localization of EGF receptor and its potential new role as a transcription factor.Nat Cell Biol 2001;3:802-808.
40.Lo HW, Hsu SC, Hung MC.EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization.Breast Cancer Res Treat 2006;95:211-218.
41.Lo HW, Xia W, Wei Y, Ali-Seyed M, Huang SF, Hung MC.Novel prognostic value of nuclear epidermal growth factor receptor in breast cancer.Cancer Res 2005;65:338-348.
42.Marti U, Burwen SJ, Wells A, Barker ME, Huling S, Feren AM, et al.Localization of epidermal growth factor receptor in hepatocyte nuclei.Hepatology 1991;13:15-20.
43.Psyrri A, Yu Z, Weinberger PM, Sasaki C, Haキy B, Camp R, et al.Quantitative determination of nuclear and cytoplasmic epidermal growth factor receptor expression in oropharyngeal squamous cell cancer by using automated quantitative analysis.Clin Cancer Res 2005;11:5856-5862.
44.Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL, et al.Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer.Mol Carcinog 2009;48:610-617.
45.Lo HW, Hung MC.Nuclear EGFR signalling network in cancers:linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival.Br J Cancer 2006;94:184-188.
46.Wang SC, Hung MC.Nuclear translocation of the epidermal growth factor receptor family membrane tyrosine kinase receptors.Clin Cancer Res 2009;15:6484-6489.
47.Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, et al.Tyrosine phosphorylation controls PCNA function through protein stability.Nat Cell Biol 2006;8:1359-1368.
48.Boerner JL, Demory ML, Silva C, Parsons SJ.Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II.Mol Cell Biol 2004;24:7059-7071.
49.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM.Structural basis for inhibition of the epidermal growth factor receptor by cetuximab.Cancer Cell 2005;7:301-311.
50.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, et al.Cetuximab Monotherapy and Cetuximab plus Irinotecan in Irinotecan-Refractory Metastatic Colorectal Cancer.N Engl J Med 2004;351:337-345.
51.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al.Platinum-Based Chemotherapy plus Cetuximab in Head and Neck Cancer.N Engl J Med 2008;359:1116-1127.
52.Yang XD, Jia XC, Corvalan JR, Wang P, Davis CG, Jakobovits A.Eradication of Established Tumors by a Fully Human Monoclonal Antibody to the Epidermal Growth Factor Receptor without Concomitant hemotherapy.Cancer Res 1999;59:1236-1243.
53.Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, et al.Open-Label Phase III Trial of Panitumumab Plus Best Supportive Care Compared With Best Supportive Care Alone in Patients With Chemotherapy-Refractory Metastatic Colorectal Cancer.J Clin Oncol 2007;25:1658-1664.
54.Mishima K, Johns TG, Luwor RB, Scott AM, Stockert E, Jungbluth AA, et al.Growth Suppression of Intracranial Xenografted Glioblastomas Overexpressing Mutant Epidermal Growth Factor Receptors by Systemic Administration of Monoclonal Antibody(mAb) 806, a Novel Monoclonal Antibody Directed to the Receptor.Cancer Res 2001;61:5349-5354.
55.Scott AM, Lee FT, Tebbutt N, Herbertson R, Gill SS, Liu Z, et al.A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors.Proc Natl Acad Sci U S A.2007;104:4071-4076.
56.Jaramillo ML, Leon Z, Grothe S, Paul-Roc B, Abulrob A, O’Connor McCourt M.Effect of the anti-receptor ligand-blocking 225 monoclonal antibody on EGF receptor endocytosis and sorting.Exp Cell Res 2006;312:2778-2790.
57.Kurai J, Chikumi H, Hashimoto K, Yamaguchi K, Yamasaki A,Sako T, et al.Antibody-Dependent Cellular Cytotoxicity Mediated by Cetuximab against Lung Cancer Cell Lines.Clin Cancer Res 2007;13:1552-1561.
58.Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK, et al.Vaccination of Malignant Glioma Patients with Peptide-pulsed Dendritic Cells Elicits Systemic Cytotoxicity and Intracranial T-cell In filtration.Cancer Res 2001;61:842-847.
59.Del Vecchio CA, Li G, Wong AJ.Targeting EGF receptor variant III: tumor-specific peptide vaccination for malignant gliomas.Expert Rev Vaccines 2012;11:133-144.
60.Zhou X, Li J, Wang Z, Chen Z, Qiu J, Zhang Y, et al.Cellular immunotherapy for carcinoma using genetically modi fied EGFR-specific T lymphocytes.Neoplasia 2013;15:544-553.
61.El-Rayes BF, LoRusso PM.Targeting the epidermal growth factor receptor.Br J Cancer 2004;91:418-424.
62.Available online: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm360574.htm(Accessed 12 Sept 2013).
63.Available online: http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/DeviceApprovalsandClearances/RecentlyApprovedDevices/ucm360819 (Accessed 12 Sept 2013).
64.Arteaga CL.Epidermal Growth Factor Receptor Dependence in Human Tumors: More Than Just Expression? Oncologist 2002;7:31-39.
65.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al.Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer.J Clin Oncol 2008;26:1626-1634.
66.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al.Immunologic Escape After Prolonged Progression-Free Survival With Epidermal Growth Factor Receptor Variant III Peptide Vaccination in Patients With Newly Diagnosed Glioblastoma.J Clin Oncol 2010;28:4722-4729.
67.Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, et al.Analysis of Epidermal Growth Factor Receptor Gene Mutation in Patients with Non??ìSmall Cell Lung Cancer and Acquired Resistance to Ge fitinib.Clin Cancer Res 2006;12:5764-5769.
68.Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al.BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models.Oncogene 2008;27:4702-4711.
69.Katakami N, Atagi S, Goto K, Hida T, Horai T, Inoue A, et al.LUX-Lung 4: A Phase II Trial of Afatinib in Patients With Advanced Non–Small-Cell Lung Cancer Who Progressed During Prior Treatment With Erlotinib, Ge fitinib, or Both.J Clin Oncol 2013;31:3335-3341.
70.Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al.MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to ge fitinib or erlotinib.Proc Natl Acad Sci U S A 2007;104:20932-20937.
71.Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T,et al.Hepatocyte Growth Factor Induces Ge fitinib Resistance of Lung Adenocarcinoma with Epidermal Growth Factor Receptor–Activating Mutations.Cancer Research 2008;68:9479-9487.
72.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C,Park JO, et al.MET Amplification Leads to Ge fitinib Resistance in Lung Cancer by Activating ERBB3 Signaling.Science 2007;316:1039-1043.
73.Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al.Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members.Oncogene 2008;27:3944-3956.
74.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al.An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as aTherapeutic Target for Overcoming EGFR Inhibitor Resistance.Clin Cancer Res 2013;19:279-290.
75.Gusenbauer S, Vlaicu P, Ullrich A.HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors.Oncogene 2013;32:3846-3856.
76.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B,Fountzilas G, et al.Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer:a retrospective consortium analysis.The Lancet Oncology 2010;11:753-762.
77.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al.Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors.N Engl J Med 2005;353:2012-2024.
78.Mellinghoff IK, Cloughesy TF, Mischel PS.PTEN-Mediated Resistance to Epidermal Growth Factor Receptor Kinase Inhibitors.Clin Cancer Res 2007;13:378-381.
79.Fenton TR, Nathanson D, Ponte de Albuquerque C, Kuga D,Iwanami A, Dang J, et al.Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the PTEN tumor suppressor at tyrosine 240.Proc Natl Acad Sci U S A 2012;109:14164-14169.
80.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al.Coactivation of Receptor Tyrosine Kinases Affects the Response of Tumor Cells to TargetedTherapies.Science 2007;318:287-290.
81.Iwanami A, Gini B, Zanca C, Matsutani T, Assuncao A, Nael A, et al.PML mediates glioblastoma resistance to mammalian target of rapamycin (mTOR)-targeted therapies.Proc Natl Acad Sci U S A 2013;110:4339-4344.
82.Lee RC, Feinbaum RL, Ambros V.The C.elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14.Cell 1993;75:843-854.
83.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T.Identification of novel genes coding for small expressed RNAs.Science 2001;294:853-858.
84.Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T.New microRNAs from mouse and human.RNA 2003;9:175-179.
85.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W,Tuschl T.Identification of tissue-specific microRNAs from mouse.Curr Biol 2002;12:735-739.
86.Vaz C, Ahmad HM, Sharma P, Gupta R, Kumar L, Kulshreshtha R, et al.Analysis of microRNA transcriptome by deep sequencing of small RNA libraries of peripheral blood.BMC Genomics 2010;11:288.
87.Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN.The Drosha-DGCR8 complex in primary microRNA processing.Genes Dev 2004;18:3016-3027.
88.Yi R, Qin Y, Macara IG, Cullen BR.Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs.Genes Dev 2003;17:3011-3016.
89.Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, et al.Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C.elegans developmental timing.Cell 2001;106:23-34.
90.Mourelatos Z, Dostie J, Paushkin S, Sharma A, Charroux B, Abel L, et al.miRNPs: a novel class of ribonucleoproteins containing numerous microRNAs.Genes Dev 2002;16:720-728.
91.Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC.The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila.Cell 2007;130:89-100.
92.Sibley CR, Seow Y, Saayman S, Dijkstra KK, El Andaloussi S, Weinberg MS, et al.The biogenesis and characterization of mammalian microRNAs of mirtron origin.Nucleic Acids Res 2012;40:438-448.
93.Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, et al.Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia.Proc Natl Acad Sci U S A 2002;99:15524-15529.
94.Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, Blanco D, et al.A microRNA DNA methylation signature for human cancer metastasis.Proc Natl Acad Sci U S A 2008;105:13556-13561.
95.Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, et al.miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis.Nat Cell Biol 2010:247-256.
96.Lee EJ, Baek M, Gusev Y, Brackett DJ, Nuovo GJ, Schmittgen TD.Systematic evaluation of microRNA processing patterns in tissues,cell lines, and tumors.RNA 2008;14:35-42.
97.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S,Miyazono K.Modulation of microRNA processing by p53.Nature 2009;460:529-533.
98.Seike M, Goto A, Okano T, Bowman ED, Schetter AJ, Horikawa I, et al.MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers.Proc Natl Acad Sci U S A 2009;106:12085-12090.
99.Huang TH, Wu F, Loeb GB, Hsu R, Heidersbach A, Brincat A, et al.Up-regulation of miR-21 by HER2/neu signaling promotes cell invasion.J Biol Chem 2009;284:18515-18524.
100.Garofalo M, Romano G, Di Leva G, Nuovo G, Jeon YJ, Ngankeu A,et al.EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and ge fitinib resistance in lung cancers.Nat Med 2011;18:74-82.
101.Talotta F, Cimmino A, Matarazzo MR, Casalino L, De Vita G,D’Esposito M, et al.An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in RAS transformation.Oncogene 2009;28:73-84.
102.Zhou X, Ren Y, Moore L, Mei M, You Y, Xu P, et al.Downregulation of miR-21 inhibits EGFR pathway and suppresses the growth of human glioblastoma cells independent of PTEN status.Lab Invest 2010;90:144-155.
103.Hatley ME, Patrick DM, Garcia MR, Richardson JA, Bassel-Duby R, van Rooij E, et al.Modulation of K-Ras-dependent lung tumorigenesis by MicroRNA-21.Cancer Cell 2010;18:282-293.
104.Ma X, Kumar M, Choudhury SN, Becker Buscaglia LE, Barker JR, Kanakamedala K, et al.Loss of the miR-21 allele elevates the expression of its target genes and reduces tumorigenesis.Proc Natl Acad Sci U S A 2011;108:10144-10149.
105.Lu Y, Liu P, Van den Bergh F, Zellmer V, James M, Wen W, et al.Modulation of gene expression and cell-cycle signaling pathways by the EGFR inhibitor ge fitinib (Iressa) in rat urinary bladder cancer.Cancer Prev Res (Phila) 2012;5:248-259.
106.Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, et al.Lin-28B transactivation is necessary for Mycmediated let-7 repression and proliferation.Proc Natl Acad Sci U S A 2009;106:3384-3389.
107.Viswanathan SR, Daley GQ, Gregory RI.Selective blockade of microRNA processing by Lin28.Science 2008;320:97-100.
108.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL,Toffanin S, et al.Lin28 promotes transformation and is associated with advanced human malignancies.Nat Genet 2009;41:843-848.
109.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, et al.RAS is regulated by the let-7 microRNA family.Cell 2005;120:635-647.
110.Li X, Carthew RW.A microRNA mediates EGF receptor signaling and promotes photoreceptor differentiation in the Drosophila eye.Cell 2005;123:1267-1277.
111.Chou YT, Lin HH, Lien YC, Wang YH, Hong CF, Kao YR, et al.EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF.Cancer Res 2010;70:8822-8831.
112.Kefas B, Godlewski J, Comeau L, Li Y, Abounader R, Hawkinson M, et al.microRNA-7 inhibits the epidermal growth factor receptor and the Akt pathway and is down-regulated in glioblastoma.Cancer Res 2008;68:3566-3572.
113.Webster RJ, Giles KM, Price KJ, Zhang PM, Mattick JS,Leedman PJ.Regulation of epidermal growth factor receptor signaling in human cancer cells by microRNA-7.J Biol Chem 2009;284:5731-5741.
114.Lee ST, Chu K, Oh HJ, Im WS, Lim JY, Kim SK, et al.Let-7 microRNA inhibits the proliferation of human glioblastoma cells.J Neurooncol 2011;102:19-24.
115.Kalinowski FC, Giles KM, Candy PA, Ali A, Ganda C, Epis MR et al.Regulation of epidermal growth factor receptor signaling and erlotinib sensitivity in head and neck cancer cells by miR-7.PLoS One 2012;7:e47067.
116.Krichevsky AM, Gabriely G.miR-21: a small multi-faceted RNA.J Cell Mol Med 2009;13:39-53.
117.Chan JA, Krichevsky AM, Kosik KS.MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells.Cancer Res 2005;65:6029-6033.
118.Frezzetti D, De Menna M, Zoppoli P, Guerra C, Ferraro A, Bello AM, et al.Upregulation of miR-21 by Ras in vivo and its role in tumor growth.Oncogene 2011;30:275-286.
119.Loayza-Puch F, Yoshida Y, Matsuzaki T, Takahashi C, Kitayama H,Noda M.Hypoxia and RAS-signaling pathways converge on, and cooperatively downregulate, the RECK tumor-suppressor protein through microRNAs.Oncogene 2010;29:2638-2648.
120.Gwak HS, Kim TH, Jo GH, Kim YJ, Kwak HJ, Kim JH, et al.Silencing of microRNA-21 confers radio-sensitivity through inhibition of the PI3K/AKT pathway and enhancing autophagy in malignant glioma cell lines.PLoS One 2012;7:e47449.
121.Hummel R, Hussey DJ, Haier J.MicroRNAs: predictors and modi fiers of chemo- and radiotherapy in different tumour types.Eur J Cancer 2010;46:298-311.
122.Cufí S, Bonavia R, Vazquez-Martin A, Oliveras-Ferraros C,Corominas-Faja B, Cuyàs E, et al.Silibinin suppresses EMT-driven erlotinib resistance by reversing the high miR-21/low miR-200c signature in vivo.Sci Rep 2013;3:2459.
123.Han M, Liu M, Wang Y, Chen X, Xu J, Sun Y, et al.Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN.PLoS One 2012;7:e39520.
124.Gong C, Yao Y, Wang Y, Liu B, Wu W, Chen J, et al.Up-regulation of miR-21 mediates resistance to trastuzumab therapy for breast cancer.J Biol Chem 2011;286:19127-19137.
125.Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, et al.Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA.Nature 2000;408:86-89.
126.Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H,Endoh H, et al.Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival.Cancer Res 2004;64:3753-3756.
127.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C et al.let-7 regulates self renewal and tumorigenicity of breast cancer cells.Cell 2007;131:1109-1123.
128.Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, et al.Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop.Nat Med 2012;18:529-537.
129.Oh JS, Kim JJ, Byun JY, Kim IA.Lin28-let7 modulates radiosensitivity of human cancer cells with activation of K-Ras.Int J Radiat Oncol Biol Phys 2010;76:5-8.
130.Ruzzo A, Graziano F, Vincenzi B, Canestrari E, Perrone G,Galluccio N, et al.High let-7a microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapy-refractory metastatic disease.Oncologist 2012;17:823-829.
131.Xia XM, Jin WY, Shi RZ, Zhang YF, Chen J.Clinical significance and the correlation of expression between Let-7 and K-ras in nonsmall cell lung cancer.Oncol Lett 2010;1:1045-1047.
132.Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, et al.A SNP in a let-7 microRNA complementary site in the KRAS 3’ untranslated region increases non-small cell lung cancer risk.Cancer Res 2008;68:8535-8540.
133.Christensen BC, Moyer BJ, Avissar M, Ouellet LG, Plaza SL,McClean MD, et al.A let-7 microRNA-binding site polymorphism in the KRAS 3’ UTR is associated with reduced survival in oral cancers.Carcinogenesis 2009;30:1003-1007.
134.Graziano F, Canestrari E, Loupakis F, Ruzzo A, Galluccio N,Santini D, et al.Genetic modulation of the Let-7 microRNA binding to KRAS 3’-untranslated region and survival of metastatic colorectal cancer patients treated with salvage cetuximabirinotecan.Pharmacogenomics J 2010;10:458-464.
135.Sebio A, Paré L, Páez D, Salazar J, González A, Sala N, et al.The LCS6 polymorphism in the binding site of let-7 microRNA to the KRAS 3’-untranslated region: its role in the efficacy of anti-EGFR-based therapy in metastatic colorectal cancer patients.Pharmacogenet Genomics 2013;23:142-147.
136.Petriella D, Galetta D, Rubini V, Savino E, Paradiso A, Simone G,et al.Molecular pro filing of thin-prep FNA samples in assisting clinical management of non-small-cell lung cancer.Mol Biotechnol 2013;54:913-919.
137.Yu Z, Ni L, Chen D, Zhang Q, Su Z, Wang Y, et al.Identification of miR-7 as an oncogene in renal cell carcinoma.J Mol Histol 2013.[Epub ahead of print].
138.Croce CM.Causes and consequences of microRNA dysregulation in cancer.Nat Rev Genet 2009;10:704-714.
139.Fang Y, Xue JL, Shen Q, Chen J, Tian L.MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma.Hepatology 2012;55:1852-1862.
140.Giles KM, Brown RA, Epis MR, Kalinowski FC, Leedman PJ.miRNA-7-5p inhibits melanoma cell migration and invasion.Biochem Biophys Res Commun 2013;430:706-710.
141.Saydam O, Senol O, Würdinger T, Mizrak A, Ozdener GB,Stemmer-Rachamimov AO, et al.miRNA-7 attenuation in Schwannoma tumors stimulates growth by upregulating three oncogenic signaling pathways.Cancer Res 2011;71:852-861.
142.Lee KM, Choi EJ, Kim IA.microRNA-7 increases radiosensitivity of human cancer cells with activated EGFR-associated signaling.Radiother Oncol 2011;101:171-176.
143.Levine AJ, Hu W, Feng Z.The P53 pathway: what questions remain to be explored? Cell Death Differ 2006;13:1027-1036.
144.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al.A microRNA component of the p53 tumour suppressor network.Nature 2007;447:1130-1134.
145.Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, et al.Transcriptional activation of miR-34a contributes to p53-mediated apoptosis.Mol Cell 2007;26:731-743.
146.Zenz T, Mohr J, Eldering E, Kater AP, Bühler A, Kienle D, et al.miR-34a as part of the resistance network in chronic lymphocytic leukemia.Blood 2009;113:3801-3808.
147.Hünten S, Siemens H, Kaller M, Hermeking H.The p53/microRNA network in cancer: experimental and bioinformatics approaches.Adv Exp Med Biol 2013;774:77-101.
148.Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A,Godwin AK, et al.Frequent downregulation of miR-34 family in human ovarian cancers.Clin Cancer Res 2010;16:1119-1128.
149.Li Y, Guessous F, Zhang Y, Dipierro C, Kefas B, Johnson E, et al.MicroRNA-34a inhibits glioblastoma growth by targeting multiple oncogenes.Cancer Res 2009;69:7569-7576.
150.Guessous F, Zhang Y, Kofman A, Catania A, Li Y, Schiff D, Purow B, et al.microRNA-34a is tumor suppressive in brain tumors and glioma stem cells.Cell Cycle 2010;9:1031-1036.
151.Migliore C, Petrelli A, Ghiso E, Corso S, Capparuccia L, Eramo A,et al.MicroRNAs impair MET-mediated invasive growth.Cancer Res 2008;68:10128-10136.
152.Zhang X, Chang A.Molecular predictors of EGFR-TKI sensitivity in advanced non-small cell lung cancer.Int J Med Sci 2008;5:209-217.
153.Hwang CI, Matoso A, Corney DC, Flesken-Nikitin A, K?rner S,Wang W, et al.Wild-type p53 controls cell motility and invasion by dual regulation of MET expression.Proc Natl Acad Sci U S A 2011;108:14240-14245.
154.Dang Y, Luo D, Rong M, Chen G.Underexpression of miR-34a in hepatocellular carcinoma and its contribution towards enhancement of proliferating inhibitory effects of agents targeting c-MET.PLoS One 2013;8:e61054.
155.Kasinski AL, Slack FJ.miRNA-34 prevents cancer initiation and progression in a therapeutically resistant K-ras and p53-induced mouse model of lung adenocarcinoma.Cancer Res 2012;72:5576-5587.
 Cancer Biology & Medicine2013年4期
Cancer Biology & Medicine2013年4期
- Cancer Biology & Medicine的其它文章
- Rare myeloid sarcoma/acute myeloid leukemia with adrenal mass after allogeneic mobilization peripheral blood stem cell transplantation
- Analysis of 30 patients with persistent or recurrent squamous cell carcinoma of the cervix within one year after concurrent chemoradiotherapy
- Effects of HLEC on the secreted proteins of epithelial ovarian cancer cells prone to metastasize to lymph nodes
- Why bortezomib cannot go with ‘green’?
- Asian trends in primary androgen depletion therapy on prostate cancer
- Translational genomics in cancer research: converting pro files into personalized cancer medicine